Analysis of the Effect of Processing Conditions on Physical Properties of Thermally Set Cellulose Hydrogels


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Preparation of Cellulose Solution
2.3. Preparation of Cellulose Hydrogels
2.4. Preparation of All-Cellulose Composite Hydrogels
2.5. Chemical Cross-Linking of Cellulose Hydrogels
2.6. Gel Analysis
2.7. Mechanical Testing
2.8. Field Emission Scanning Electron Microscopy (FE-SEM)
2.9. Pore Size Analysis
2.10. Cross-Linking Efficiency
2.11. Fourier Transform Infrared (FTIR) Spectroscopy
2.12. Manufacture of Cellulose Gels with Complex Geometric Features
3. Results and Discussion
3.1. Manufacture of Cellulose Hydrogels with Complex Structure
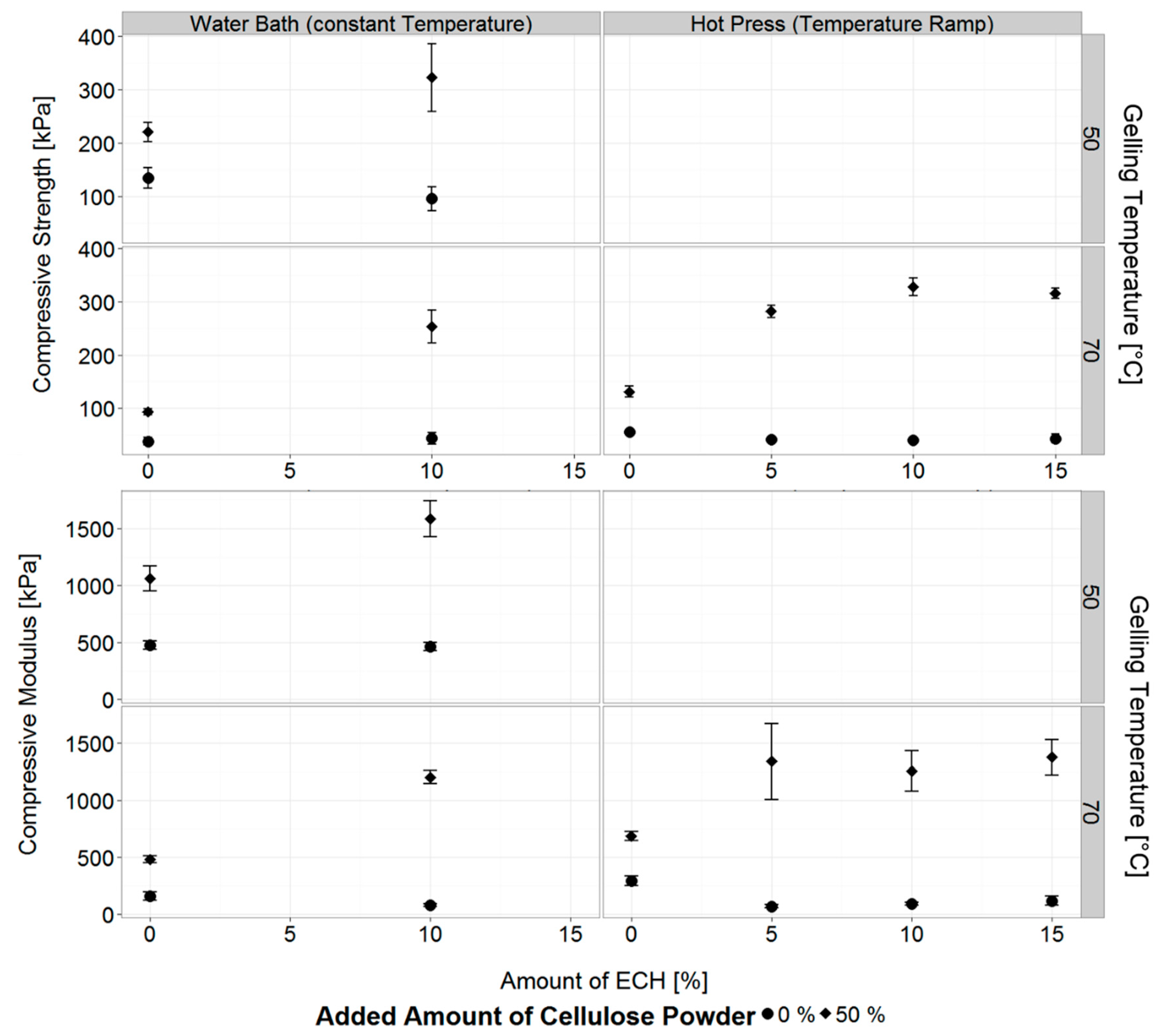
3.2. Influence of Temperature, Gelling Method and Physical Cross-Linker on Mechanical Properties of Cellulose Hydrogels
3.3. Properties of Chemically Cross-Linked Cellulose Hydrogels
3.3.1. Concurrent Chemical Cross-Linking and Gelation of Cellulose Hydrogels
3.3.2. Post Cross-Linking of Cellulose Hydrogels
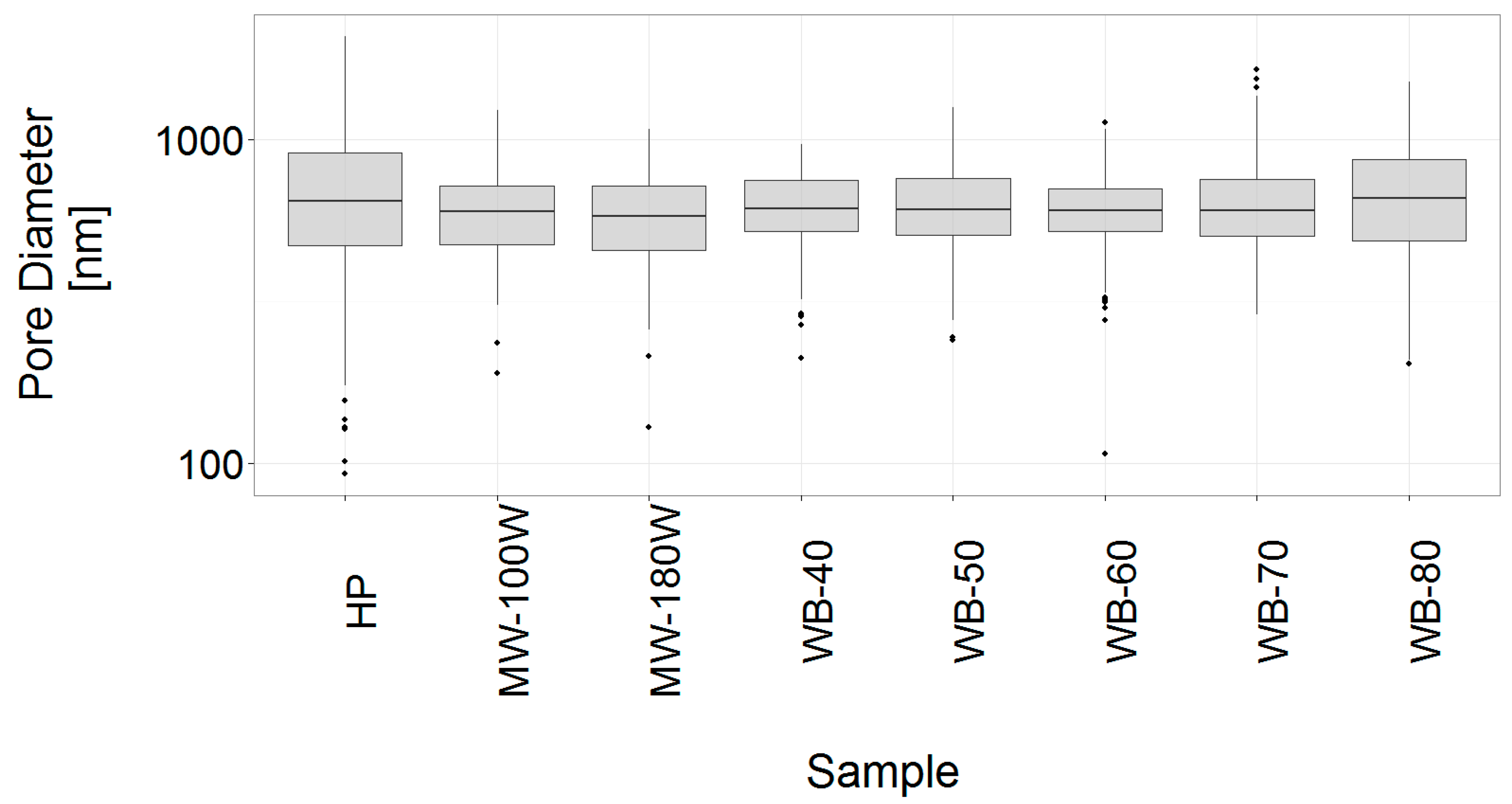
3.4. Analysis of the Gel Microstructure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chang, C.; Zhang, L. Cellulose-based hydrogels: Present status and application prospects. Carbohyd. Polym. 2011, 84, 40–53. [Google Scholar] [CrossRef]
- Peppas, N.; Huang, Y.; Torres-Lugo, M.; Ward, J.; Zhang, J. Physicochemical foundations and structural design of hydrogels in medicine and biology. Annu. Rev. Biomed. Eng. 2000, 2, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Klemm, D.; Heublein, B.; Fink, H.P.; Bohn, A. Cellulose: Fascinating biopolymer and sustainable raw material. Angew. Chem. Int. Edit. 2005, 44, 3358–3393. [Google Scholar] [CrossRef]
- Gericke, M.; Trygg, J.; Fardim, P. Functional cellulose beads: Preparation, characterization, and applications. Chem. Rev. 2013, 113, 4812–4836. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Luo, Q.; Zhou, D. Affinity membrane chromatography for the analysis and purification of proteins. J. Biochem. Bioph. Meth. 2001, 49, 199–240. [Google Scholar] [CrossRef] [Green Version]
- Gericke, M.; Schlufter, K.; Liebert, T.; Heinze, T.; Budtova, T. Rheological properties of cellulose/ionic liquid solutions: From dilute to concentrated states. Biomacromolecules 2009, 10, 1188–1194. [Google Scholar] [CrossRef]
- Fink, H.-P.; Weigel, P.; Purz, H.; Ganster, J. Structure formation of regenerated cellulose materials from NMMO-solutions. Prog. Polym. Sci. 2001, 26, 1473–1524. [Google Scholar] [CrossRef]
- Conio, G.; Corazza, P.; Bianchi, E.; Tealdi, A.; Ciferri, A. Phase equilibria of cellulose in N, N-dimethylacetamide/LiCl solutions. J. Polym. Sci. Pol. Lett. 1984, 22, 273–277. [Google Scholar] [CrossRef]
- Kosan, B.; Michels, C.; Meister, F. Dissolution and forming of cellulose with ionic liquids. Cellulose 2008, 15, 59–66. [Google Scholar] [CrossRef]
- Pinkert, A.; Marsh, K.N.; Pang, S.; Staiger, M.P. Ionic liquids and their interaction with cellulose. Chem. Rev. 2009, 109, 6712–6728. [Google Scholar] [CrossRef] [PubMed]
- Isogai, A.; Atalla, R. Dissolution of cellulose in aqueous NaOH solutions. Cellulose 1998, 5, 309–319. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, L.; Cai, J. Behavior of cellulose in NaOH/urea aqueous solution characterized by light scattering and viscometry. J. Polym. Sci. Pol. Phys. 2004, 42, 347–353. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, L.; Chang, C.; Cheng, G.; Chen, X.; Chu, B. Hydrogen-Bond-Induced Inclusion Complex in Aqueous Cellulose/LiOH/Urea Solution at Low Temperature. ChemPhysChem 2007, 8, 1572–1579. [Google Scholar] [CrossRef]
- Xiong, B.; Zhao, P.; Hu, K.; Zhang, L.; Cheng, G. Dissolution of cellulose in aqueous NaOH/urea solution: Role of urea. Cellulose 2014, 21, 1183–1192. [Google Scholar] [CrossRef]
- Isobe, N.; Noguchi, K.; Nishiyama, Y.; Kimura, S.; Wada, M.; Kuga, S. Role of urea in alkaline dissolution of cellulose. Cellulose 2013, 20, 97–103. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, L. Rapid dissolution of cellulose in LiOH/urea and NaOH/urea aqueous solutions. Macromol. Biosci. 2005, 5, 539–548. [Google Scholar] [CrossRef]
- Gavillon, R.; Budtova, T. Aerocellulose: New highly porous cellulose prepared from cellulose- naoh aqueous solutions. Biomacromolecules 2007, 9, 269–277. [Google Scholar] [CrossRef]
- Budtova, T.; Navard, P. Cellulose in NaOH-water based solvents: A review. Cellulose 2016, 23, 5–55. [Google Scholar] [CrossRef]
- Huber, T.; Clucas, D.; Vilmay, M.; Pupkes, B.; Stuart, J.; Dimartino, S.; Fee, C. 3D Printing Cellulose Hydrogels Using LASER Induced Thermal Gelation. J. Manuf. Mater. Process. 2018, 2, 42. [Google Scholar] [CrossRef]
- Fee, C.; Gordon, A.; Huber, T.; Dimartino, S. Triply periodic minimal surface structures in 3D-printed chromatography columns. In Proceedings of the 30th International Symposium on Preparative and Process Chromatography, Philadelphia, PA, USA, 16–19 July 2017. [Google Scholar]
- Torres-Rendon, J.G.; Köpf, M.; Gehlen, D.; Blaeser, A.; Fischer, H.; Laporte, L.D.; Walther, A. Cellulose Nanofibril Hydrogel Tubes as Sacrificial Templates for Freestanding Tubular Cell Constructs. Biomacromolecules 2016, 17, 905–913. [Google Scholar] [CrossRef]
- Jia, N.; Li, S.-M.; Ma, M.-G.; Sun, R.-C.; Zhu, J.-F. Hydrothermal synthesis and characterization of cellulose-carbonated hydroxyapatite nanocomposites in NaOH-urea aqueous solution. Sci. Adv. Mater. 2010, 2, 210–214. [Google Scholar] [CrossRef]
- Jia, N.; Li, S.-M.; Zhu, J.-F.; Ma, M.-G.; Xu, F.; Wang, B.; Sun, R.-C. Microwave-assisted synthesis and characterization of cellulose-carbonated hydroxyapatite nanocomposites in NaOH-urea aqueous solution. Mater. Let. 2010, 64, 2223–2225. [Google Scholar] [CrossRef]
- Sannino, A.; Demitri, C.; Madaghiele, M. Biodegradable cellulose-based hydrogels: Design and applications. Materials 2009, 2, 353–373. [Google Scholar] [CrossRef]
- Lenzi, F.; Sannino, A.; Borriello, A.; Porro, F.; Capitani, D.; Mensitieri, G. Probing the degree of crosslinking of a cellulose based superabsorbing hydrogel through traditional and NMR techniques. Polymer 2003, 44, 1577–1588. [Google Scholar] [CrossRef]
- Rodríguez, R.; Alvarez-Lorenzo, C.; Concheiro, A. Interactions of ibuprofen with cationic polysaccharides in aqueous dispersions and hydrogels: Rheological and diffusional implications. Eur. J. Pharm. Sci. 2003, 20, 429–438. [Google Scholar] [CrossRef]
- Sannino, A.; Madaghiele, M.; Conversano, F.; Mele, G.; Maffezzoli, A.; Netti, P.; Ambrosio, L.; Nicolais, L. Cellulose derivative-hyaluronic acid-based microporous hydrogels cross-linked through divinyl sulfone (DVS) to modulate equilibrium sorption capacity and network stability. Biomacromolecules 2004, 5, 92–96. [Google Scholar] [CrossRef]
- Huber, T.; Müssig, J.; Curnow, O.; Pang, S.; Bickerton, S.; Staiger, M.P. A critical review of all-cellulose composites. J. Mater. Sci. 2012, 47, 1171–1186. [Google Scholar] [CrossRef]
- Loth, F.; Philipp, B. The macroporous network of “bead cellulose” and its response to crosslinking with epichlorohydrin. Makromol. Chem. Macromol. Symp. 1989, 30, 273–287. [Google Scholar] [CrossRef]
- McKelvey, J.B.; Benerito, R.R.; Berni, R.J.; Hattox, C.A. The cellulose-epichlorohydrin reaction in the presence of neutral salt and salt-alkali solutions. Text. Res. J. 1964, 34, 759–767. [Google Scholar] [CrossRef]
- McKelvey, J.B.; Webre, B.G.; Klein, E. Reaction of epoxides with cotton cellulose in the presence of sodium hydroxide. Text. Res. J. 1959, 29, 918–925. [Google Scholar] [CrossRef]
- Bai, Y.-X.; Li, Y.-F. Preparation and characterization of crosslinked porous cellulose beads. Carbohyd. Polym. 2006, 64, 402–407. [Google Scholar] [CrossRef]
- Oliveira, W.D.; Glasser, W.G. Hydrogels from polysaccharides. I. Cellulose beads for chromatographic support. J. Appl. Polym. Sci. 1996, 60, 63–73. [Google Scholar] [CrossRef]
- Qin, X.; Lu, A.; Zhang, L. Gelation behavior of cellulose in NaOH/urea aqueous system via cross-linking. Cellulose 2013, 20, 1669–1677. [Google Scholar] [CrossRef]
- Hockaday, L.A.; Kang, K.H.; Colangelo, N.W.; Cheung, P.Y.C.; Duan, B.; Malone, E.; Wu, J.; Girardi, L.N.; Bonassar, L.J.; Lipson, H.; et al. Rapid 3D printing of anatomically accurate and mechanically heterogeneous aortic valve hydrogel scaffolds. Biofabrication 2012, 4, 035005. [Google Scholar] [CrossRef]
- Torres-Rendon, J.G.; Femmer, T.; De Laporte, L.; Tigges, T.; Rahimi, K.; Gremse, F.; Zafarnia, S.; Lederle, W.; Ifuku, S.; Wessling, M. Others Bioactive gyroid scaffolds formed by sacrificial templating of nanocellulose and nanochitin hydrogels as instructive platforms for biomimetic tissue engineering. Adv. Mater. 2015, 27, 2989–2995. [Google Scholar] [CrossRef]
- Qi, H.; Chang, C.; Zhang, L. Effects of temperature and molecular weight on dissolution of cellulose in NaOH/urea aqueous solution. Cellulose 2008, 15, 779–787. [Google Scholar] [CrossRef]
- Ruan, D.; Lue, A.; Zhang, L. Gelation behaviors of cellulose solution dissolved in aqueous NaOH/thiourea at low temperature. Polymer 2008, 49, 1027–1036. [Google Scholar] [CrossRef]
- Jin, H.; Nishiyama, Y.; Wada, M.; Kuga, S. Nanofibrillar cellulose aerogels. Colloids Surf. A 2004, 240, 63–67. [Google Scholar] [CrossRef]
- Savina, I.N.; Ingavle, G.C.; Cundy, A.B.; Mikhalovsky, S.V. A simple method for the production of large volume 3D macroporous hydrogels for advanced biotechnological, medical and environmental applications. Sci. Rep. 2016, 6, 21154. [Google Scholar] [CrossRef] [Green Version]
- Kim, U.J.; Park, J.; Li, C.Y.; Jin, H.Y.; Valluzzi, R.; Kaplan, D. Structure and Properties of Silk Hydrogels. Biomacromolecules 2004, 5, 786–792. [Google Scholar] [CrossRef]
- Kim, S.H.; Chu, C.C. Pore structure analysis of swollen dextran-methacrylate hydrogels by SEM and mercury intrusion porosimetry. J. Biomed. Mater. Res. 2000, 53, 258–266. [Google Scholar] [CrossRef]
- Van Vlierberghe, S.; Cnudde, V.; Dubruel, P.; Masschaele, B.; Cosijns, A.; De Paepe, I.; Jacobs, P.J.; Van Hoorebeke, L.; Remon, J.P.; Schacht, E. Porous gelatin hydrogels: 1. Cryogenic formation and structure analysis. Biomacromolecules 2007, 8, 331–337. [Google Scholar] [CrossRef]
- Karcher, H. The triply periodic minimal surfaces of Alan Schoen and their constant mean curvature companions. Manuscripta Math. 1989, 64, 291–357. [Google Scholar] [CrossRef]
- Schoen, A.H. Infinite Periodic Minimal Surfaces without Self-Intersections; NASA: Washington, DC, USA, 1970.
- Dolamore, F. In Silico Analysis of Flow and Dispersion in Ordered Porous Media. Ph.D. Thesis, University of Canterbury, Christchurch, New Zealand, 2017. [Google Scholar]
- Dimartino, S.; Fee, C.; Nawada, S.; Clucas, D.; Huber, T.; Gordon, A.; Dolamore, F. 3D Printing of Porous Media at the Microstructural Scale; University of Canterbury: Christchurch, New Zealand, 2014. [Google Scholar]
- Ek, R.; Lennholm, H.; Davidson, R.; Nyström, C.; Ragnarsson, G. Pore swelling in beads made of cellulose fibres and fibre fragments. Int. J. Pharm. 1995, 122, 49–56. [Google Scholar] [CrossRef]
- Haas, D.W.; Hrutfiord, B.F.; Sarkanen, K. Kinetic study on the alkaline degradation of cotton hydrocellulose. J. Appl. Polym. Sci. 1967, 11, 587–600. [Google Scholar] [CrossRef]
- Al-Nasassrah, M.A.; Podczeck, F.; Newton, J.M. The effect of an increase in chain length on the mechanical properties of polyethylene glycols. Eur. J. Pharm. Biopharm. 1998, 46, 31–38. [Google Scholar] [CrossRef]
- Kappe, C.O. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Edit. 2004, 43, 6250–6284. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, L. Impacts of nanowhisker on formation kinetics and properties of all-cellulose composite gels. Carbohyd. Polym. 2011, 83, 1937–1946. [Google Scholar] [CrossRef]
- Delval, F.; Crini, G.; Bertini, S.; Filiatre, C.; Torri, G. Preparation, characterization and sorption properties of crosslinked starch-based exchangers. Carbohyd. Polym. 2005, 60, 67–75. [Google Scholar] [CrossRef]
- Guo, B.; Chen, W.; Yan, L. Preparation of flexible, highly transparent, cross-linked cellulose thin film with high mechanical strength and low coefficient of thermal expansion. ACS Sustain. Chem. Eng. 2013, 1, 1474–1479. [Google Scholar] [CrossRef]
- Lee, S.-H.; Park, S.-Y.; Choi, J.-H. Fiber formation and physical properties of chitosan fiber crosslinked by epichlorohydrin in a wet spinning system: The effect of the concentration of the crosslinking agent epichlorohydrin. J. Appl. Polym. Sci. 2004, 92, 2054–2062. [Google Scholar] [CrossRef]
- Zhou, J.; Chang, C.; Zhang, R.; Zhang, L. Hydrogels prepared from unsubstituted cellulose in NaOH/urea aqueous solution. Macromol. Biosci. 2007, 7, 804–809. [Google Scholar] [CrossRef]
- Grdadolnik, J.; Maréchal, Y. Urea and urea-water solutions—An infrared study. J. Mol. Struct. 2002, 615, 177–189. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts; John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Dimartino, S.; Savory, D.M.; Fraser-Miller, S.J.; Gordon, K.C.; McQuillan, A.J. Microscopic and infrared spectroscopic comparison of the underwater adhesives produced by germlings of the brown seaweed species Durvillaea antarctica and Hormosira banksii. J. R. Soc. Interface 2016, 13, 20151083. [Google Scholar] [CrossRef] [Green Version]
- Dehabadi, L.; Wilson, L.D. Polysaccharide-based materials and their adsorption properties in aqueous solution. Carbohyd. Polym. 2014, 113, 471–479. [Google Scholar] [CrossRef]
- Udoetok, I.A.; Dimmick, R.M.; Wilson, L.D.; Headley, J.V. Adsorption properties of cross-linked cellulose-epichlorohydrin polymers in aqueous solution. Carbohyd. Polym. 2016, 136, 329–340. [Google Scholar] [CrossRef]
- Mariano-Torres, J.A.; López-Marure, A.; Domiguez-Sánchez, M.Á. Synthesis and characterization of polymers based on citric acid and glycerol: Its application in non-biodegradable polymers. Dyna 2015, 82, 53–59. [Google Scholar] [CrossRef]
- Kuniak, L.; Marchessault, R. Study of the crosslinking reaction between epichlorohydrin and starch. Starch-Stärke 1972, 24, 110–116. [Google Scholar] [CrossRef]
- Luby, P.; Kuniak, L.; Fanter, C. Crosslinking statistics, 3. Relation between relative reactivity and accessibility of cellulose hydroxyl groups. Die Makromol. Chem. 1979, 180, 2379–2386. [Google Scholar] [CrossRef]
- Barrangou, L.M.; Daubert, C.R.; Allen Foegeding, E. Textural properties of agarose gels. I. Rheological and fracture properties. Food Hydrocoll. 2006, 20, 184–195. [Google Scholar] [CrossRef]
- Normand, V.; Lootens, D.L.; Amici, E.; Plucknett, K.P.; Aymard, P. New insight into agarose gel mechanical properties. Biomacromolecules 2000, 1, 730–738. [Google Scholar] [CrossRef]
- Cao, X.; Yu, F.; Li, Y.; Zeng, L.; Zhu, J.; Wang, G.; Chen, X. Diels-Alder crosslinked HA/PEG hydrogels with high elasticity and fatigue resistance for cell encapsulation and articular cartilage tissue repair. Polym. Chem. 2014, 5, 5116–5123. [Google Scholar]
- Yang, J.; Han, C.; Xu, F.; Sun, R. Simple approach to reinforce hydrogels with cellulose nanocrystals. Nanoscale 2014, 6, 5934–5943. [Google Scholar] [CrossRef]
- Hoepfner, S.; Ratke, L.; Milow, B. Synthesis and characterisation of nanofibrillar cellulose aerogels. Cellulose 2008, 15, 121–129. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huber, T.; Feast, S.; Dimartino, S.; Cen, W.; Fee, C. Analysis of the Effect of Processing Conditions on Physical Properties of Thermally Set Cellulose Hydrogels. Materials 2019, 12, 1066. https://doi.org/10.3390/ma12071066
Huber T, Feast S, Dimartino S, Cen W, Fee C. Analysis of the Effect of Processing Conditions on Physical Properties of Thermally Set Cellulose Hydrogels. Materials. 2019; 12(7):1066. https://doi.org/10.3390/ma12071066
Chicago/Turabian StyleHuber, Tim, Sean Feast, Simone Dimartino, Wanwen Cen, and Conan Fee. 2019. "Analysis of the Effect of Processing Conditions on Physical Properties of Thermally Set Cellulose Hydrogels" Materials 12, no. 7: 1066. https://doi.org/10.3390/ma12071066