Degradation Performance of Open-Cell Biomaterials from Phosphated Carbonyl Iron Powder with PEG Coating

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Fe Foam Preparation
2.2. Fe/P Foam Preparation
2.3. Preparation of Polymer Coating Layer
2.4. Material Characterization
2.5. Electrochemical Corrosion Test
2.5.1. Potentiodynamic Polarization Tests
2.5.2. Electrochemical Impedance Spectroscopy
2.6. Immersion Corrosion Test
3. Results and Discussion
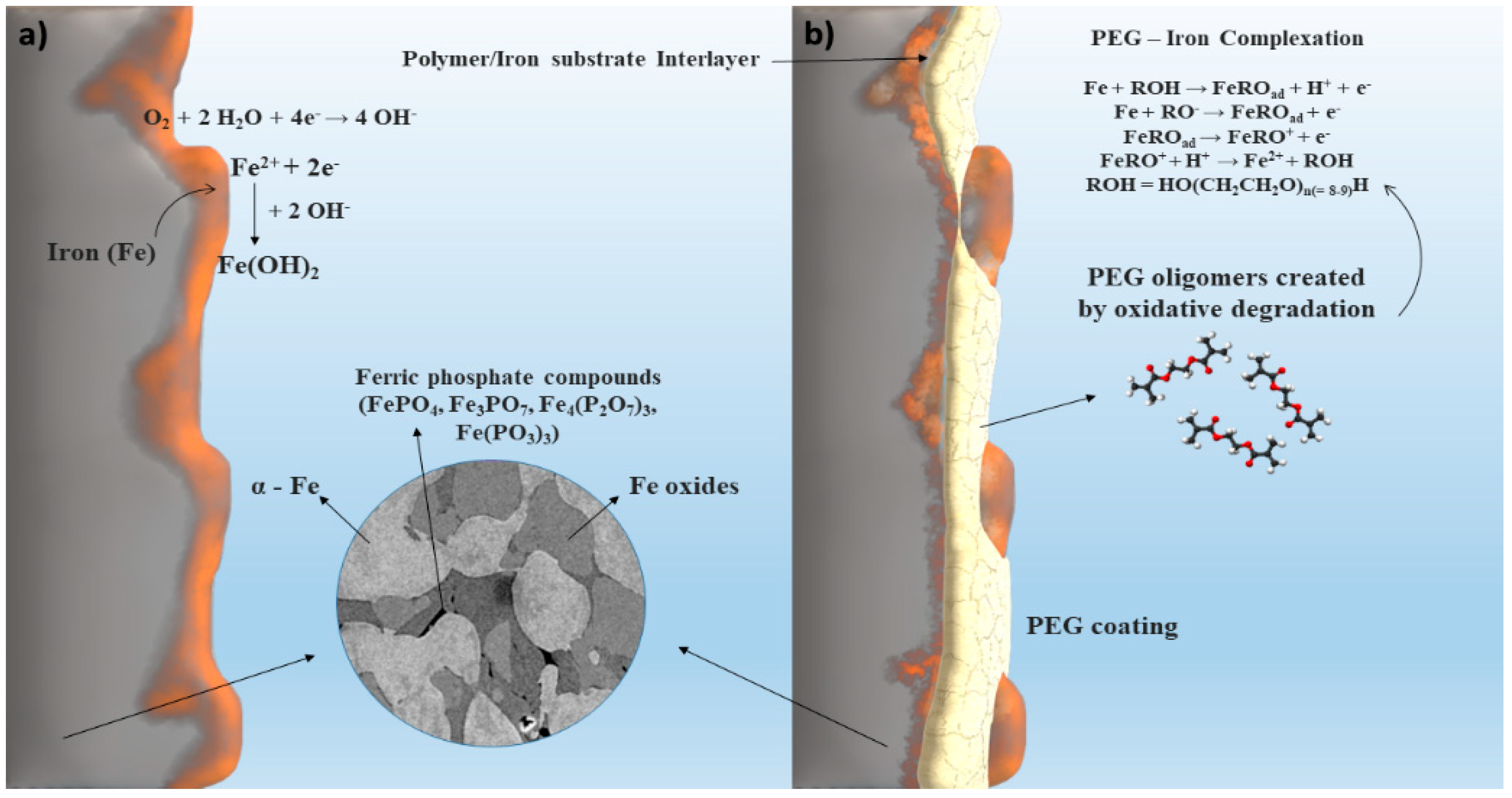
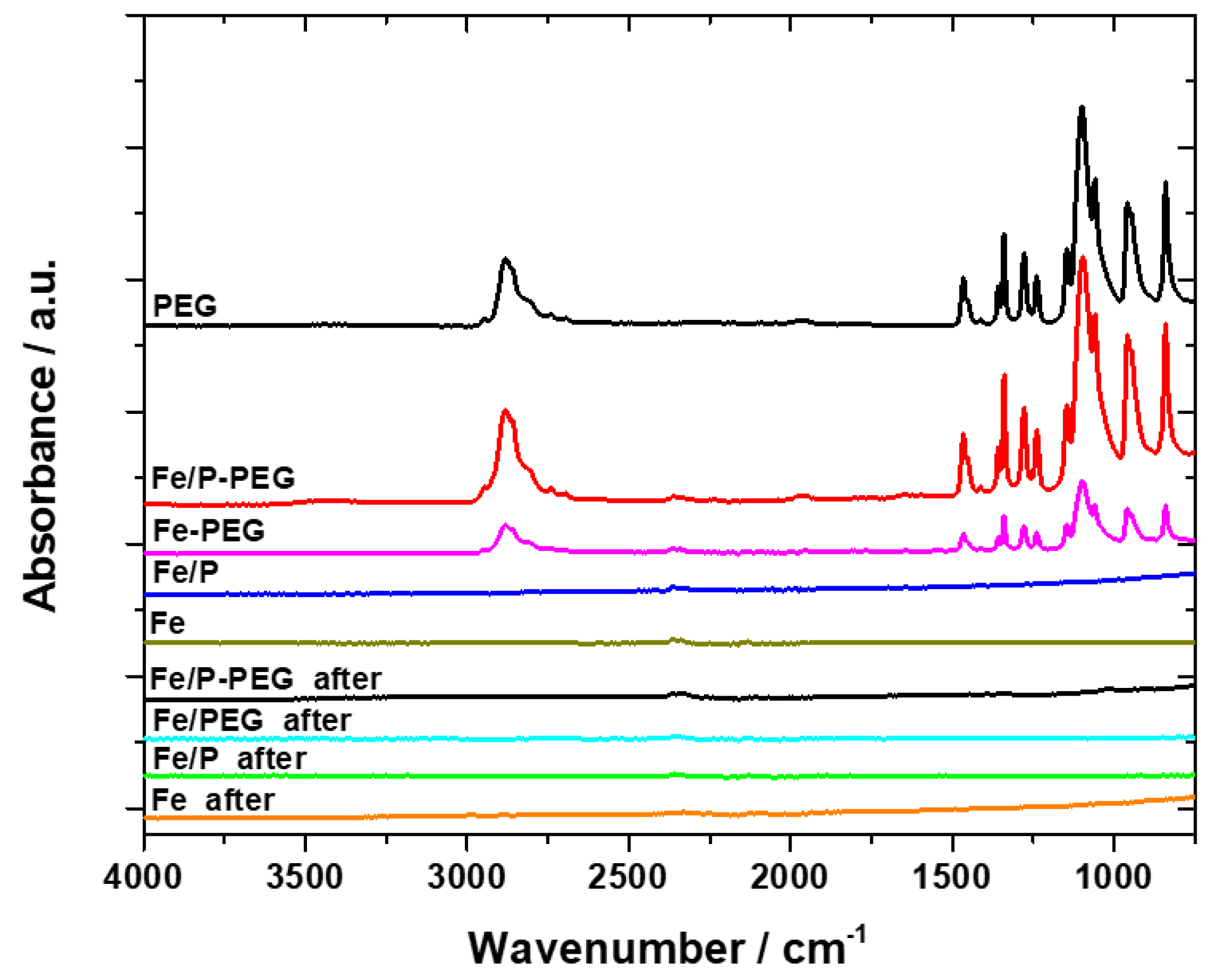
3.1. Surface Morphology and Composition
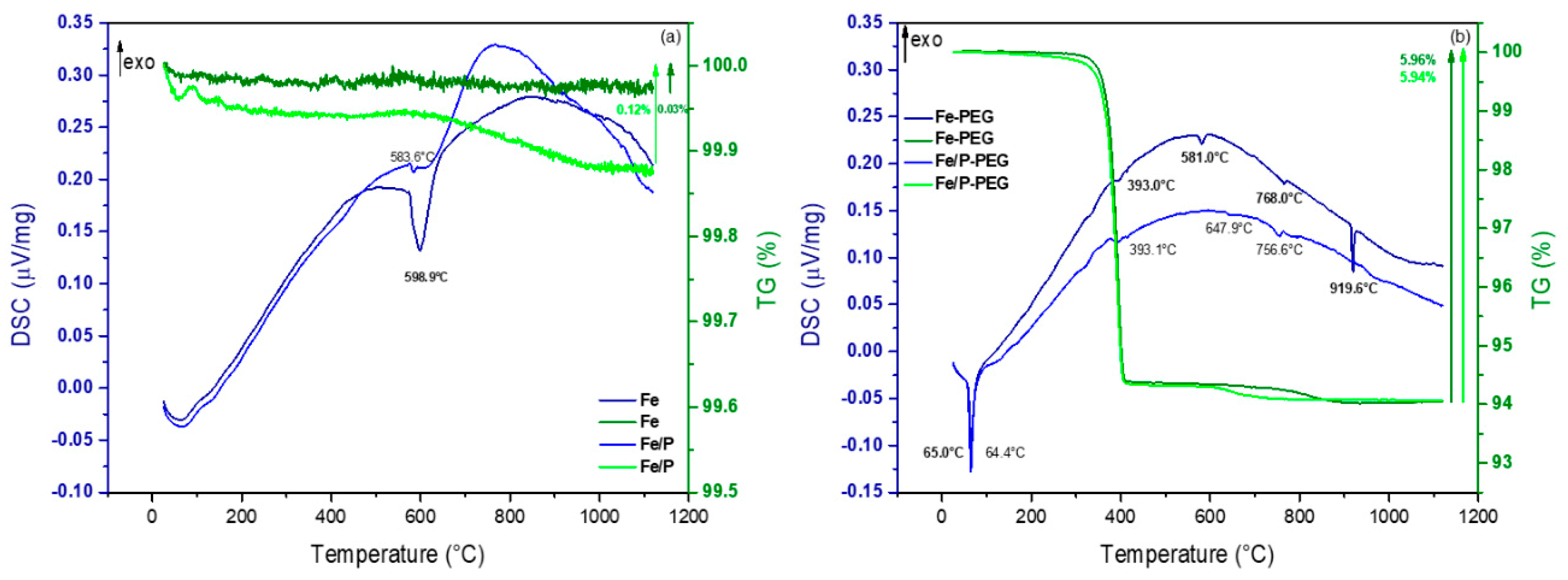
3.2. Thermogravimetric Analysis
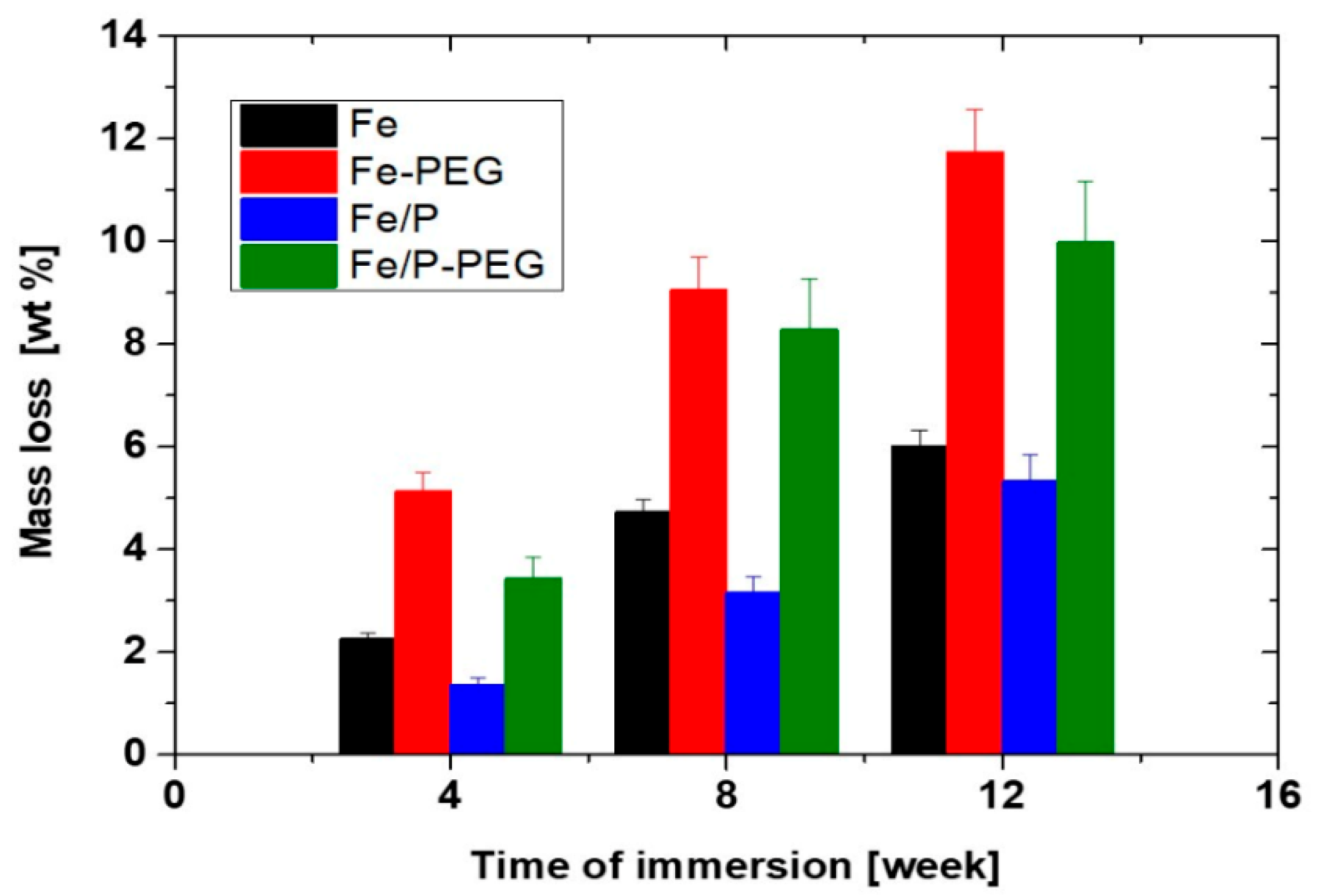
3.3. Immersion Corrosion Behavior
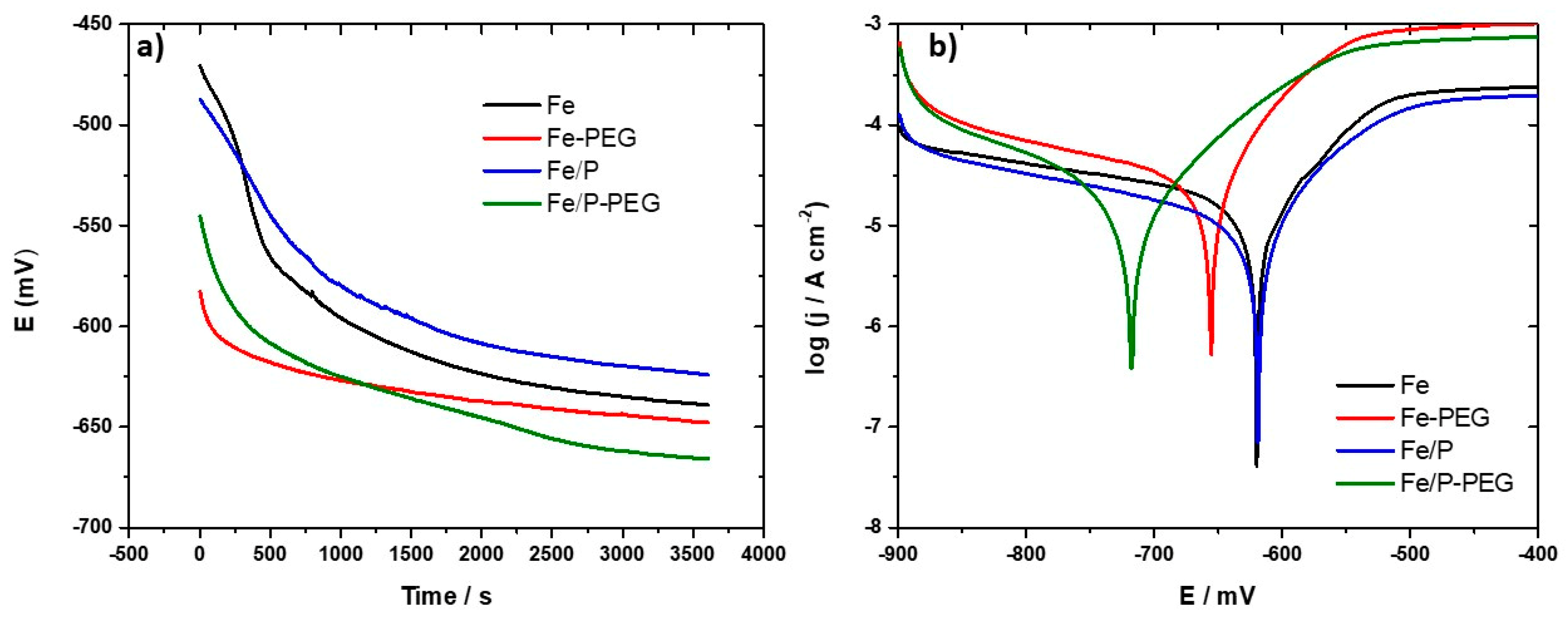
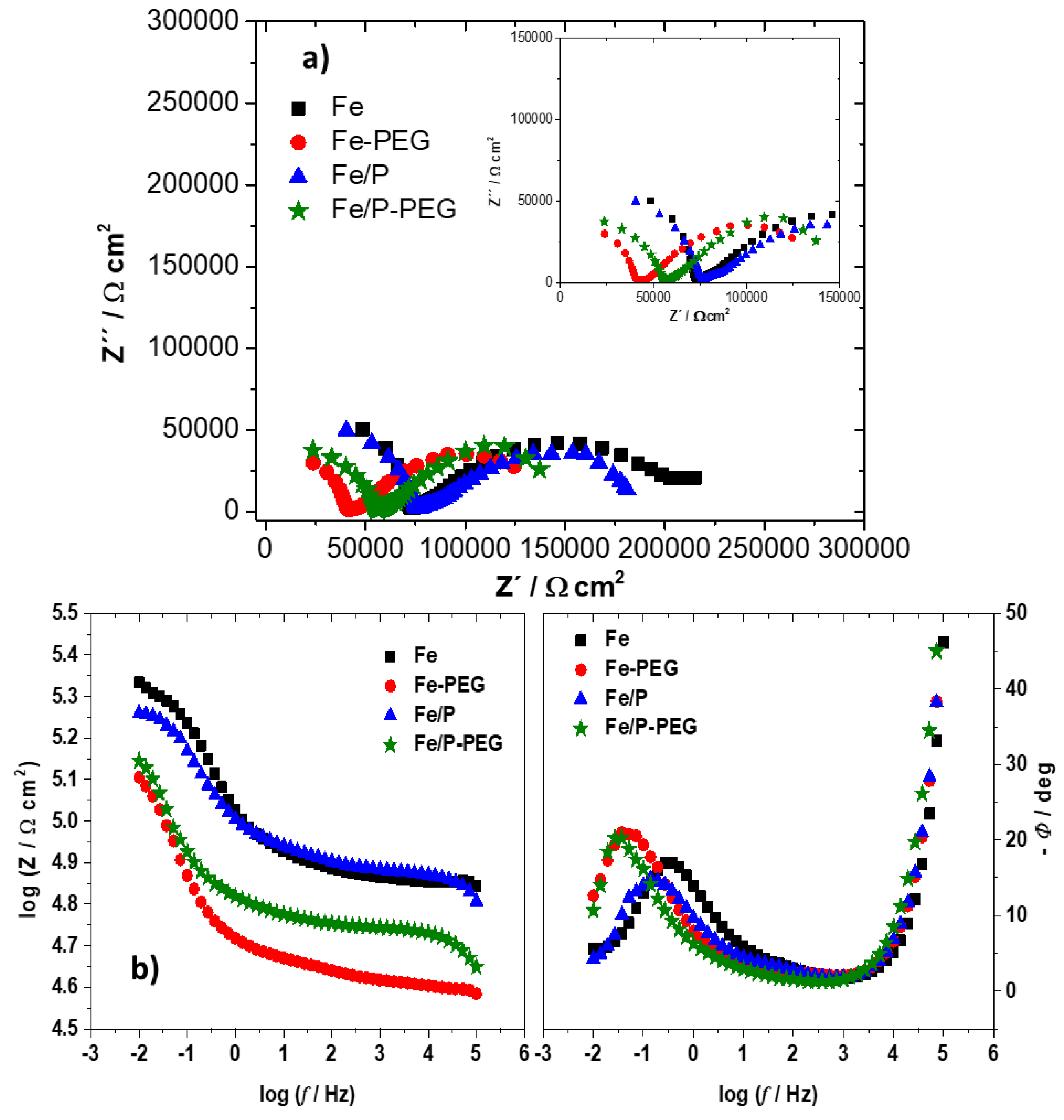
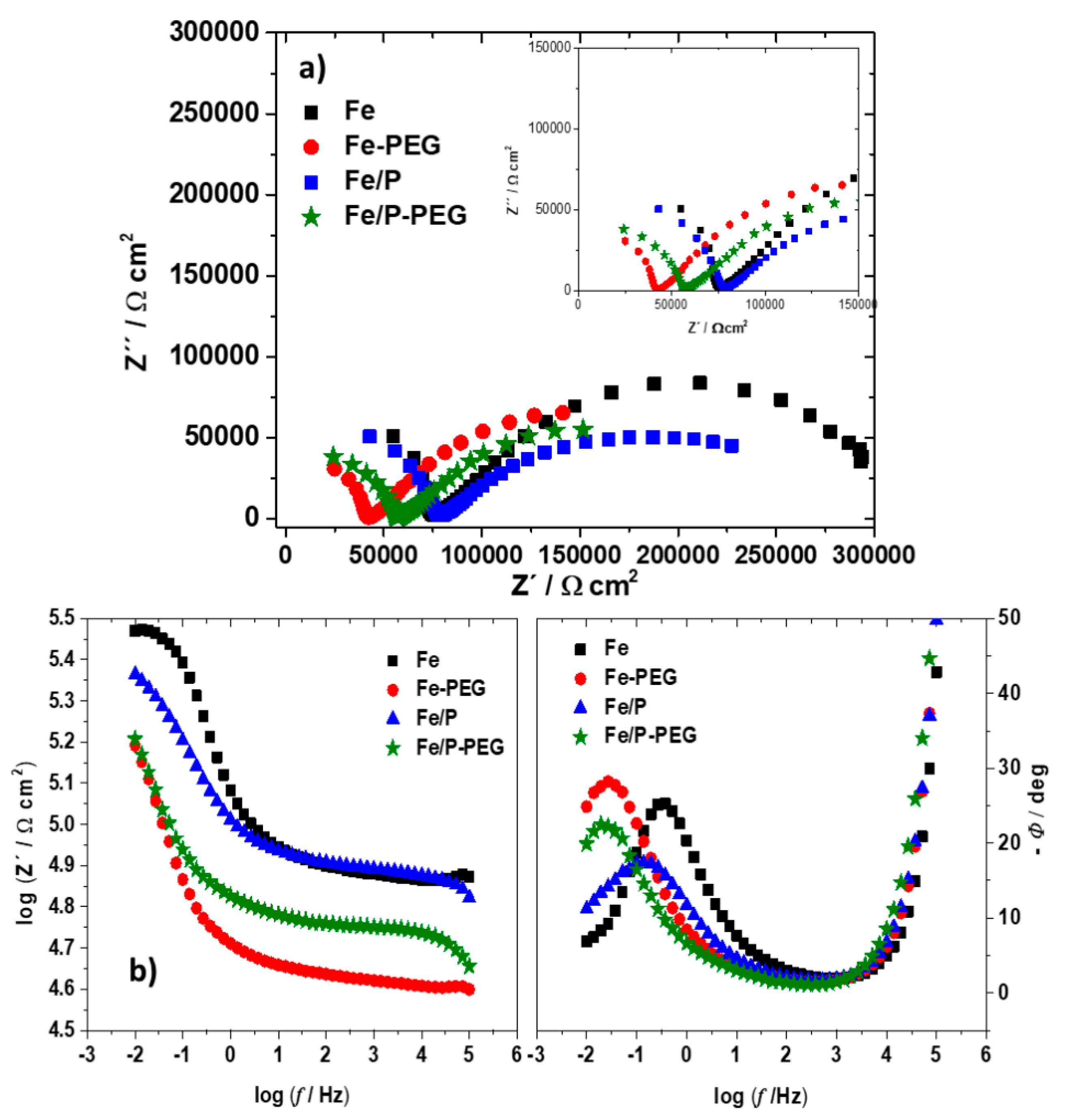
3.4. Electrochemical Corrosion Behavior
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, H.; Zheng, Y.; Qin, L. Progress of biodegradable metals, Review. Prog. Nat. Sci. Mater. Int. 2014, 24, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Purnama, A.; Hermawan, H.; Couet, J.; Mantovani, D. Assessing the biocompatibility of degradable metallic materials: State-of-the-art and focus on the potential of genetic regulation. Acta Biomater. 2015, 6, 1800–1807. [Google Scholar] [CrossRef] [PubMed]
- Prakasam, M.; Locs, J.; Salma-Ancane, K.; Loca, D.; Largeteau, A.; Berzina-Cimdina, L. Biodegradable Materials and Metallic Implants—A Review. J. Funct. Biomater. 2017, 8, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Xu, Y.; Zhou, J.; Li, H.; Chang, J.; Huan, Z. In vitro degradation and surface bioactivity of iron-matrix composites containing silicate-based bioceramic. Bioact. Mater. 2017, 2, 10–18. [Google Scholar] [CrossRef]
- Heiden, M.; Walker, E.; Stanciu, L. Magnesium, Iron and Zinc Alloys, the Trifecta of Bioresorbable Orthopaedic and Vascular Implantation—A Review. J. Biotechnol. Biomater. 2015, 5, 1000178. [Google Scholar]
- Ulum, M.F.; Arafat, A.; Noviana, D.; Yusop, A.H.; Nasution, A.K.; Kadir, M.A.; Hermawan, H. In vitro and in vivo degradation evaluation of novel iron-bioceramic composites for bone implant applications. Mater. Sci. Eng. C 2014, 36, 336–344. [Google Scholar] [CrossRef]
- Hermawan, H. Updates on the research and development of absorbable metals for biomedical applications. Prog. Biomater. 2018, 7, 93–110. [Google Scholar] [CrossRef] [Green Version]
- Radha, R.; Sreekanth, D. Insight of magnesium alloys and composites for orthopedic implant applications—A review. J. Magnes. Alloy. 2017, 5, 286–312. [Google Scholar] [CrossRef]
- Sharma, P.; Pandey, P.M. Corrosion behaviour of the porous iron scaffold in simulated body fluid for biodegradable implant application. Mater. Sci. Eng. C 2019, 99, 838–852. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, N.; Zhu, D. Endothelial Cellular Responses to Biodegradable Metal Zinc. ACS Biomater. Sci. Eng. 2015, 1, 1174–1182. [Google Scholar] [CrossRef] [Green Version]
- Yusop, A.H.; Bakir, A.A.; Shaharom, N.A.; Abdul Kadir, M.R.; Hermawan, H. Porous BiodegradableMetals for Hard Tissue Scaffolds: A Review. Int. J. Biomater. 2012, 2012, 641430. [Google Scholar] [CrossRef] [Green Version]
- Čapek, J.; Vojtěch, D.; Oborná, A. Microstructural and mechanical properties of biodegradable iron foam prepared by powder metallurgy. Mater. Des. 2015, 83, 468–482. [Google Scholar] [CrossRef]
- Sharma, P.; Pandey, P.M.M. Morphological and mechanical characterization of topologically ordered open cell porous iron foam fabricated using 3D printing and pressureless microwave sintering. Mater. Des. 2018, 160, 442–454. [Google Scholar] [CrossRef]
- Wegener, B.; Sievers, B.; Utzschneider, S.; Müller, P.; Jansson, V.; Rößler, S.; Quadbeck, P. Microstructure, cytotoxicity and corrosion of powder-metallurgical iron alloys for biodegradable bone replacement materials. Mater. Sci. Eng. B 2011, 176, 1789–1796. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, J.; Pavanram, P.; Leeflang, M.A.; Fockaert, L.I.; Pouran, B.; Tümer, N.; Schröder, K.U.; Mol, J.M.C.; Weinans, H.; et al. Additively manufactured biodegradable porous magnesium. Acta Biomater. 2018, 67, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Ulum, M.F.; Caesarendra, W.; Alavi, R.; Hermawan, H. In-Vivo Corrosion Characterization and Assessment. Coatings 2019, 9, 282. [Google Scholar] [CrossRef] [Green Version]
- Hermawan, H.; Alamdari, H.; Mantovani, D.; Dube, D. Iron–manganese: New class of metallic degradable biomaterials prepared by powder metallurgy. Powder Metall. 2008, 51, 38–45. [Google Scholar] [CrossRef]
- Yusop, A.H.M.; Daud, N.M.; Nur, H.; Kadir, M.R.A.; Hermawan, H. Controlling the degradation kinetics of porous iron by poly (lactic-co-glycolic acid) infiltration for use as temporary medical implants. Sci. Rep. 2015, 15, 11194. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, Z.; Frank, M.A.; Detsch, R.; Boccaccini, A.R.; Virtanen, S. Accelerated Degradation Behavior and Cytocompatibility of Pure Iron Treated with Sandblasting. ACS Appl. Mater. Interfaces 2016, 8, 26482–26492. [Google Scholar] [CrossRef]
- Noviana, D.; Estuningsih, S.; Paramitha, D.; Ulum, M.F.; Hermawan, H. In-vitro Cytotoxicity and In-vivo Tissue Response Study of Foreign Bodies Iron Based Materials. Adv. Mat. Res. 2015, 1112, 449–452. [Google Scholar] [CrossRef]
- Cheng, J.; Liu, B.; Wu, Y.H.; Zheng, Y.F. Comparative in vitro Study on Pure Metals (Fe, Mn, Mg, Zn and W) as Biodegradable Metals. J. Mater. Sci. Technol. 2013, 29, 619–627. [Google Scholar] [CrossRef]
- Stevens, M.M. Biomaterials for bone tissue engineering. Mater. Today 2008, 11, 18–25. [Google Scholar] [CrossRef]
- Lefevre, L.-P.; Banhart, J.; Dunand, D.C. Porous Metals and Metallic Foams: Current Status and Recent Developments. Adv. Eng. Mater. 2008, 10, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Champagne, S.; Trenggono, A.; Tolouei, R.; Mantovani, D.; Hermawan, H. Development and characterization of silver containing calcium phosphate coatings on pure iron foam intended for bone scaffold applications. Mater. Des. 2018, 148, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Haverová, L.; Oriňaková, R.; Oriňak, A.; Gorejová, R.; Baláž, M.; Vanýsek, P.; Kupková, M.; Hrubovčáková, M.; Mudroň, P.; Radoňák, J.; et al. An In Vitro Corrosion Study of Open Cell Iron Structures with PEG Coating for Bone Replacement Applications. Metals 2018, 8, 499. [Google Scholar] [CrossRef] [Green Version]
- Oriňaková, R.; Gorejová, R.; Macko, J.; Oriňak, A.; Kupková, M.; Hrubovčáková, M.; Ševc, J.; Smith, R.M. Evaluation of In Vitro Biocompatibility of Open Cell Iron Structures with PEG Coating. Appl. Surf. Sci. 2019, 475, 515–518. [Google Scholar] [CrossRef]
- Orinakova, R.; Orinak, A.; Giretova, M.; Medvecky, L.; Kupkova, M.; Hrubovcakova, M.; Maskalova, I.; Macko, J.; Kalavsky, F. A study of cytocompatibility and degradation of iron-based biodegradable materials. J. Biomater. Appl. 2016, 30, 1060–1070. [Google Scholar] [CrossRef]
- Oriňaková, R.; Gorejová, R.; Orságová Králová, Z.; Haverová, L.; Oriňak, A.; Maskaľová, I.; Kupková, M.; Džupon, M.; Baláž, M.; Hrubovčáková, M.; et al. Evaluation of mechanical properties and hemocompatibility of open cell iron foams with polyethylene glycol coating. Appl. Surf. Sci. 2020, 505, 144634. [Google Scholar] [CrossRef]
- Hrubovčáková, M.; Kupková, M.; Džupon, M. Fe and Fe-P Foam for Biodegradable Bone Replacement Material: Morphology, Corrosion Behaviour, and Mechanical Properties. Adv. Mater. Sci. Eng. 2016, 2016, 6257368. [Google Scholar] [CrossRef] [Green Version]
- ASTM G59. Standard Test Method for Conducting Potentiodynamic Polarization Resistance Measurements; ASTM: West Conshohocken, PA, USA, 2009. [Google Scholar]
- ASTM G31. Standard Guide for Laboratory Immersion Corrosion Testing of Metals; ASTM: West Conshohocken, PA, USA, 2012. [Google Scholar]
- Kou, Y.; Wang, S.; Luo, J.; Sun, K.; Zhang, J.; Tan, Z.; Shi, Q. Thermal analysis and heat capacity study of polyethylene glycol (PEG) phase change materials for thermal energy storage applications. J. Chem. Thermodyn. 2019, 128, 259–274. [Google Scholar] [CrossRef]
- Pielichowski, K.; Flejtuch, K. Non-oxidative thermal degradation of poly(ethylene oxide): Kinetic and thermoanalytical study. J. Anal. Appl. Pyrolysis 2005, 73, 131–138. [Google Scholar] [CrossRef]
- Han, S.; Kim, C.; Kwon, D. Thermal/oxidative degradation and stabilization of polyethylene glycol. Polymer 1997, 38, 317–323. [Google Scholar] [CrossRef]
- Dudrová, E.; Kabátová, M.; Hvizdoš, P.; Oriňaková, R. Sintered composite materials on the basis of Fe/FePO4-coated powders. Surf. Interface Anal. 2015, 47, 350–356. [Google Scholar] [CrossRef]
- Noor, E.A.; Al-Moubarak, A.H. Corrosion behavior of mild steel in hydrochloric acid solutions. Int. J. Electrochem. Sci. 2008, 3, 806–818. [Google Scholar]
- Chen, B.; Evans, J.R.G.; Holding, S. Decomposition of Poly(ethylene glycol) in Nanocomposites. J. Appl. Polym. Sci. 2004, 94, 548–552. [Google Scholar] [CrossRef]
- Wu, J.; Lu, X.; Tan, L.; Zhang, B.; Yang, K. Effect of hydrion evolution by polylactic-co-glycolic acid coating on degradation rate of pure iron. J. Biomed. Mater. Res. B 2013, 101, 1222–1232. [Google Scholar] [CrossRef]
- Wang, Y.B.; Li, H.F.; Zheng, Y.F.; Li, M. Corrosion performances in simulated body fluids and cytotoxicity evaluation of Fe-based bulk metallic glasses. Mater. Sci. Eng. C 2012, 32, 599–606. [Google Scholar] [CrossRef]
- Ribeiro, D.V.; Souza, C.A.C.; Abrantes, J.C.C. Use of Electrochemical Impedance Spectroscopy (EIS) to monitoring the corrosion of reinforced concrete. Ibracon Struct. Mater. J. 2015, 8, 529–546. [Google Scholar] [CrossRef]
- Cheng, Q.; Chen, Z. The Cause Analysis of the Incomplete Semi-Circle Observed in High Frequency Region of EIS Obtained from TEL-Covered Pure Copper. Int. J. Electrochem. Sci. 2013, 8, 8282–8290. [Google Scholar]
- Shameli, K.; Ahmad, M.B.; Jazayeri, S.D.; Sedaghat, S.; Shabanzadeh, P.; Jahangirian, H.; Mahdavi, M.; Abdollahi, Y. Synthesis and Characterization of Polyethylene Glycol Mediated Silver Nanoparticles by the Green Method. Int. J. Mol. Sci. 2012, 13, 6639–6650. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | ρ (g·cm−3) | πT (%) | πO (%) | πC (%) | PS (μm) |
|---|---|---|---|---|---|
| Fe | 0.72 | 85 | 80 | 5 | 400–1500 |
| Fe-PEG | 0.95 | 73 | 61 | 12 | 350–1000 |
| Fe/P | 0.81 | 80 | 74 | 6 | 400–1700 |
| Fe/P-PEG | 0.97 | 70 | 61 | 9 | 350–1100 |
| Sample | Chemical Composition (wt.%) | |||
|---|---|---|---|---|
| Fe | C | O | P | |
| Fe | 100 | 0 | 0 | 0 |
| Fe-PEG | 25.51 | 46.12 | 28.37 | 0 |
| Fe/P | 99.53 | 0 | 0 | 0.47 |
| Fe/P-PEG | 5.38 | 53.87 | 40.75 | 0 |
| Time of Immersion (Weeks) | CR (mm·Year−1) | |||
|---|---|---|---|---|
| Fe | Fe-PEG | Fe/P | Fe/P-PEG | |
| 4 | 0.0229 ± 0.0014 | 0.0492 ± 0.0020 | 0.0136 ± 0.0015 | 0.0415 ± 0.0031 |
| 8 | 0.0231 ± 0.0018 | 0.0455 ± 0.0036 | 0.0131 ± 0.0021 | 0.0397 ± 0.0025 |
| 12 | 0.0222 ± 0.0017 | 0.0398 ± 0.0028 | 0.0102 ± 0.0018 | 0.0364 ± 0.0022 |
| Sample | Ecorr (mV) | jcorr (μA·cm−2) | CR (mm·Year−1) |
|---|---|---|---|
| Fe | −620 | 16.33 | 0.200 |
| Fe-PEG | −658 | 29.72 | 0.362 |
| Fe/P | −617 | 9.80 | 0.151 |
| Fe/P-PEG | −718 | 18.65 | 0.280 |
| Sample | Before Immersion | After Immersion | ||
|---|---|---|---|---|
| Rct (kΩ·cm2) | Cdl (μF·cm−1) | Rct (kΩ·cm2) | Cdl (μF·cm−1) | |
| Fe | 146.6 ± 1.7 | 530.6 ± 6.1 | 232.1 ± 2.8 | 671.3 ± 7.8 |
| Fe-PEG | 89.28 ± 0.6 | 155.4 ± 1.3 | 117.1 ± 1.3 | 153.5 ± 1.7 |
| Fe/P | 105.5 ± 1.5 | 320.3 ± 4.0 | 156.8 ± 1.6 | 396.7 ± 5.0 |
| Fe/P-PEG | 87.91 ± 0.9 | 127.4 ± 1.1 | 110.3 ± 1.3 | 129.0 ± 1.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oriňaková, R.; Gorejová, R.; Petráková, M.; Králová, Z.O.; Oriňak, A.; Kupková, M.; Hrubovčáková, M.; Podobová, M.; Baláž, M.; Smith, R.M. Degradation Performance of Open-Cell Biomaterials from Phosphated Carbonyl Iron Powder with PEG Coating. Materials 2020, 13, 4134. https://doi.org/10.3390/ma13184134
Oriňaková R, Gorejová R, Petráková M, Králová ZO, Oriňak A, Kupková M, Hrubovčáková M, Podobová M, Baláž M, Smith RM. Degradation Performance of Open-Cell Biomaterials from Phosphated Carbonyl Iron Powder with PEG Coating. Materials. 2020; 13(18):4134. https://doi.org/10.3390/ma13184134
Chicago/Turabian StyleOriňaková, Renáta, Radka Gorejová, Martina Petráková, Zuzana Orságová Králová, Andrej Oriňak, Miriam Kupková, Monika Hrubovčáková, Mária Podobová, Matej Baláž, and Roger M. Smith. 2020. "Degradation Performance of Open-Cell Biomaterials from Phosphated Carbonyl Iron Powder with PEG Coating" Materials 13, no. 18: 4134. https://doi.org/10.3390/ma13184134