First–Principles Investigation of the Structural, Elastic, Electronic, and Optical Properties of α– and β–SrZrS3: Implications for Photovoltaic Applications


Abstract
:
1. Introduction
2. Computational Details
3. Results and Discussion
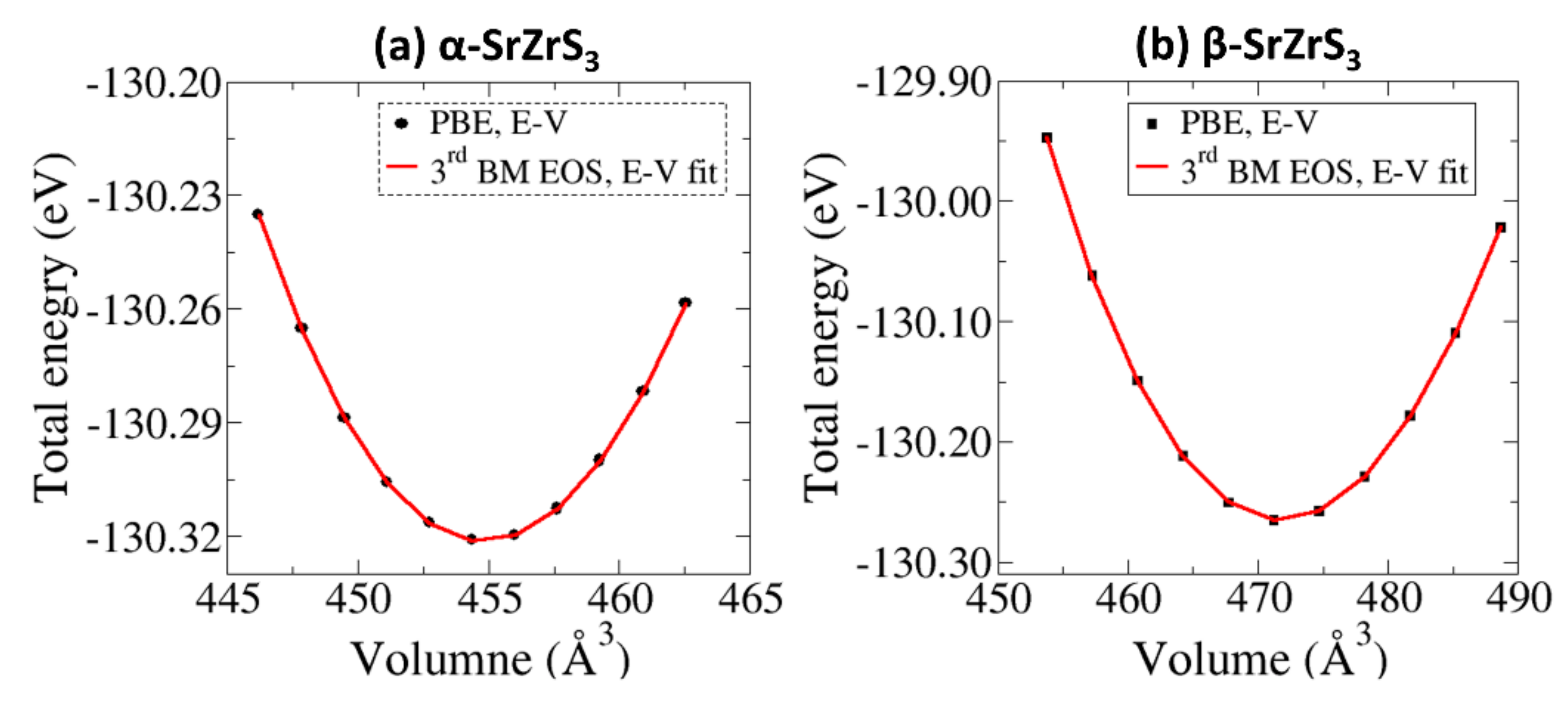
3.1. Structural Properties
3.2. Mechanical Properties
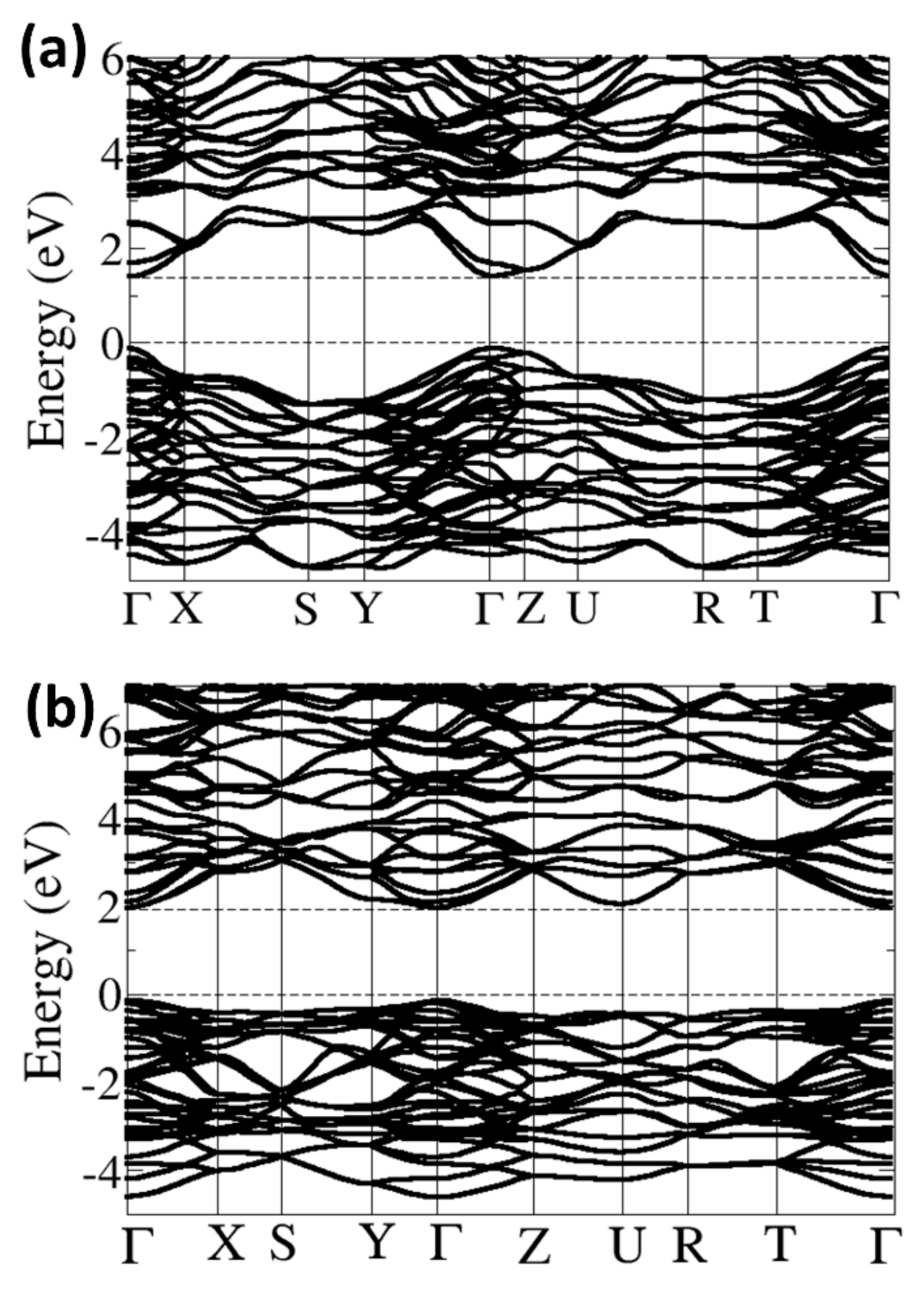
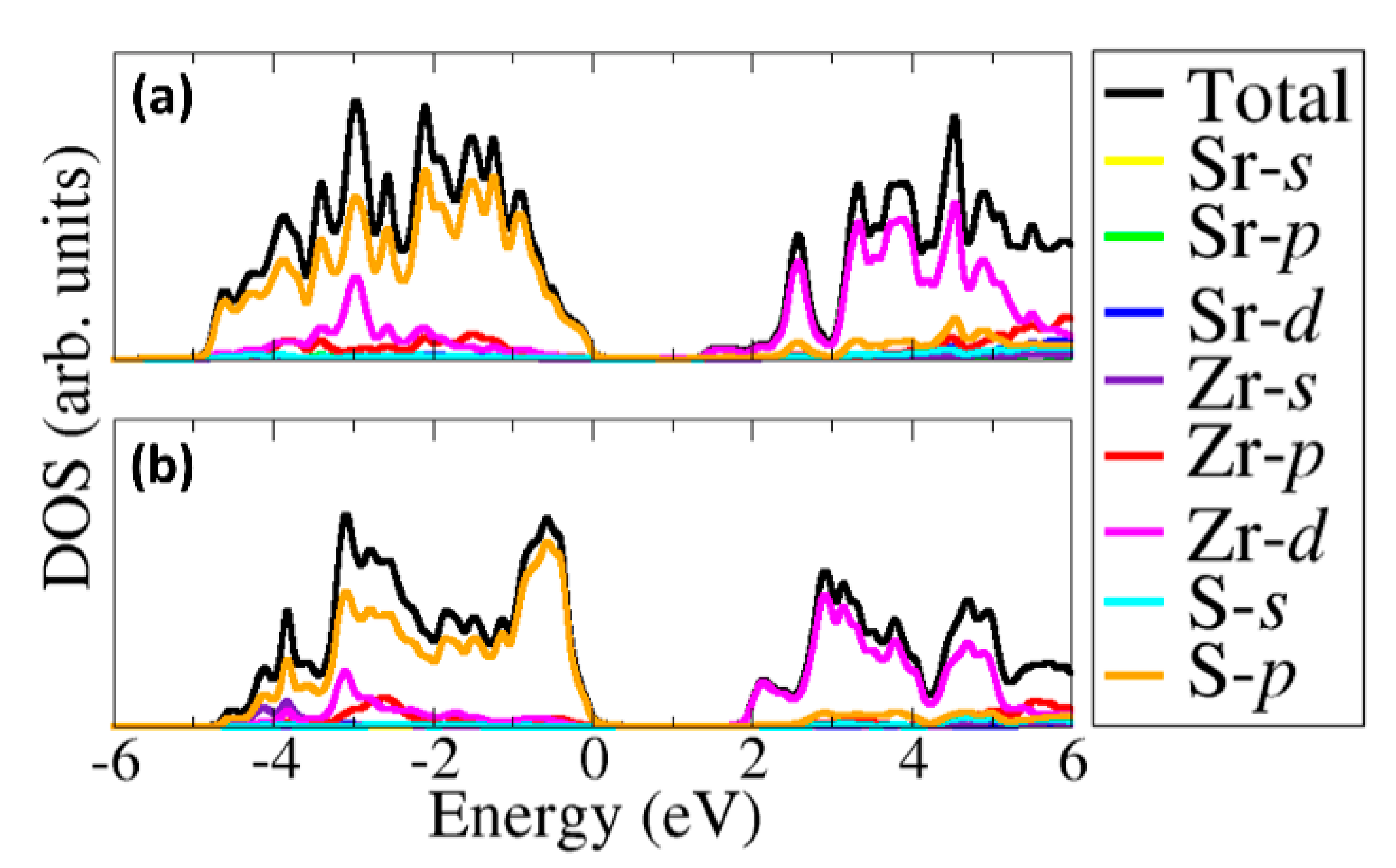
3.3. Electronic Properties
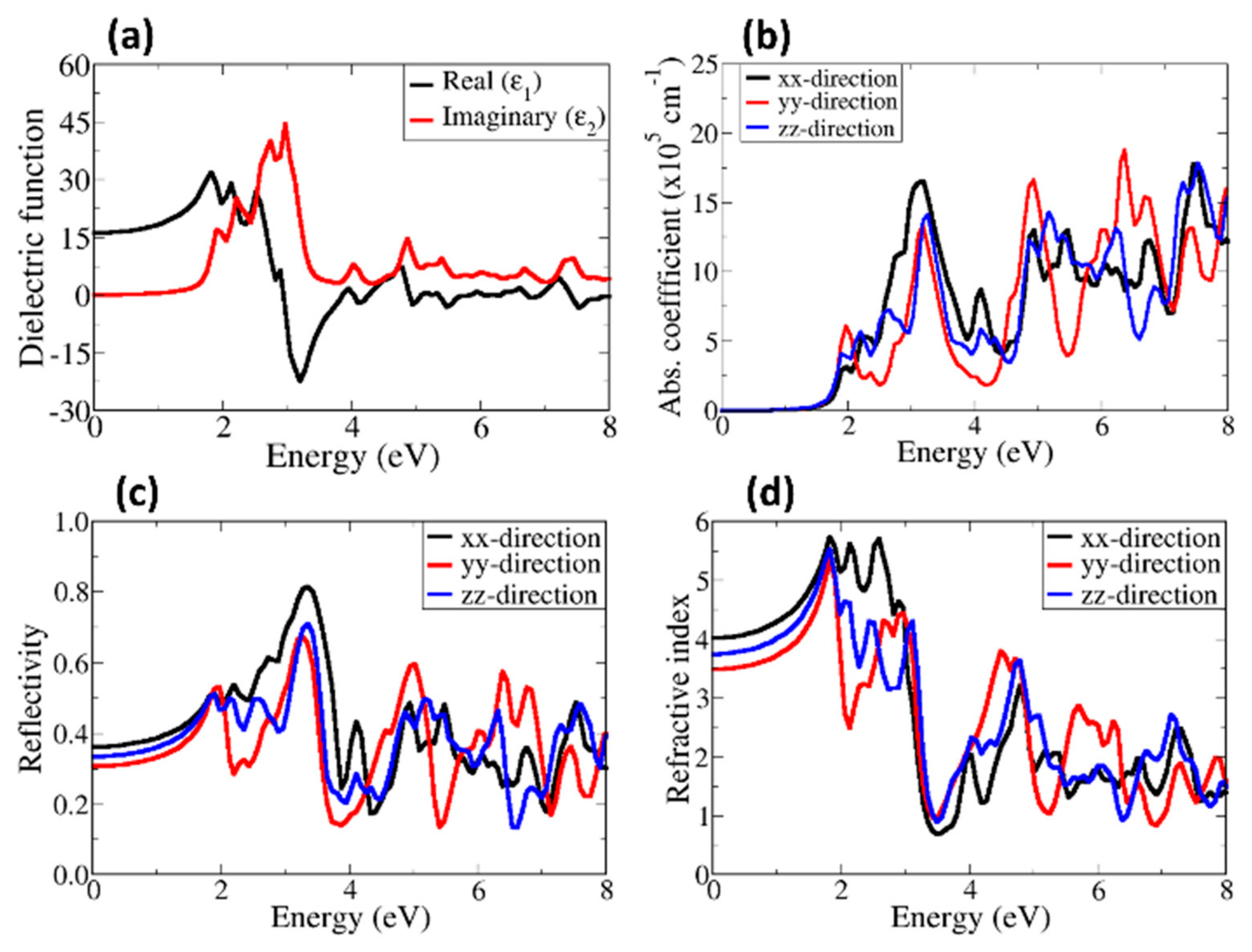
3.4. Optical Properties
4. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kojima, A.; Teshima, K.; Shirai, Y.; Miyasaka, T. Organometal Halide Perovskites as Visible–Light Sensitizers for Photovoltaic Cells. J. Am. Chem. Soc. 2009, 131, 6050–6051. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.J.; Shang, S.L.; Du, Y.; Kong, Y.; Zhang, L.J.; Chen, L.; Zhao, D.D.; Liu, Z.K. Planar Heterojunction Perovskite Solar Cells via Vapor–Assisted Solution Process. J. Am. Chem. Soc. 2014, 136, 622–625. [Google Scholar]
- Sun, Y.; Agiorgousis, M.L.; Zhang, P.; Zhang, S. Chalcogenide Perovskites for Photovoltaics. Nano Lett. 2015, 15, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Noel, N.K.; Stranks, S.D.; Abate, A.; Wehrenfennig, C.; Guarnera, S.; Haghighirad, A.; Sadhanala, A.; Eperon, G.E.; Pathak, S.K.; Johnston, M.B.; et al. Lead–free organic–inorganic tin halide perovskites for photovoltaic applications. Energy Environ. Sci. 2014, 7, 3061–3068. [Google Scholar] [CrossRef]
- Shuyan, S.; Liu, J.; Portale, G.; Fang, H.; Blake, G.R.; Brink, G.H.; Koster, L.A.; Loi, M.A. Highly Reproducible Sn-Based Hybrid Perovskite Solar Cells with 9% Efficiency. Adv. Energy Mater. 2018, 8, 1702019. [Google Scholar]
- Song, T.; Yokoyama, T.; Aramaki, S.; Kanatzidis, M.G. Performance Enhancement of Lead–Free Tin–based Perovskite Solar Cells with Reducing Atmosphere– Assisted Dispersible Additive. ACS Energy Lett. 2017, 24, 897–903. [Google Scholar] [CrossRef]
- Yang, B.; Chen, J.; Yang, S.; Hong, F.; Sun, L.; Han, P.; Pullerits, T.; Deng, W.; Han, K. Lead–Free Silver–Bismuth Halide Double Perovskite Nanocrystals. Angew. Chem. Int. Ed. Engl. 2018, 4, 5359–5363. [Google Scholar] [CrossRef]
- Sanders, S.; Stümmler, D.; Pfeiffer, P.; Ackermann, N.; Simkus, G.; Heuken, M.; Baumann, P.K.; Vescan, A.; Kalisch, H. Chemical Vapor Deposition of Perovskite Films for Solar Cell Application. Sci. Rep. 2019, 9, 9774. [Google Scholar] [CrossRef]
- Durrant, J.R. Green fabrication of stable lead–free bismuth based perovskite solar cells using a non–toxic solvent. Commun. Chem. 2019, 2, 1–7. [Google Scholar]
- Chen, M.; Ju, M.; Carl, A.D.; Zong, Y.; Grimm, R.L.; Gu, J.; Zeng, X.C.; Zhou, Y.; Padture, N. Cesium Titanium (IV) Bromide Thin Films Based Stable Lead–free Perovskite Solar Cells. Joule 2018, 2, 558–570. [Google Scholar] [CrossRef] [Green Version]
- Nechache, R.; Harnagea, C.; Li, S.; Cardenas, L.; Huang, W.; Chakrabartty, J.; Rosei, F. Bandgap tuning of multiferroic oxide solar cells. Nat. Photon. 2015, 9, 61–67. [Google Scholar] [CrossRef]
- Grinberg, I.; West, D.V.; Torres, M.; Gou, G.; Stein, D.M.; Wu, L.; Chen, G.; Gallo, E.M.; Akbashev, A.R.; Davies, P.K.; et al. Perovskite oxides for visible–light–absorbing ferroelectric and photovoltaic materials. Nature 2013, 503, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Saparov, B.; Hong, F.; Wang, J.; Mitzi, D.B.; Yan, Y. Alloying and Defect Control within Chalcogenide Perovskites for Optimized Photovoltaic Application Alloying and Defect Control within Chalcogenide Perovskites for Optimized Photovoltaic Application. Chem. Mater. 2016, 283, 821–829. [Google Scholar] [CrossRef]
- Ju, M.; Dai, J.; Ma, L.; Zeng, X.C. Perovskite Chalcogenides with Optimal Bandgap and Desired Optical Absorption for Photovoltaic Devices. Adv. Energy Mater. 2017, 8, 2–8. [Google Scholar] [CrossRef]
- Oumertem, M.; Saadi, D.M.; Rai, N.B.D.P.; Ibrir, R.K.M. Theoretical investigation of the structural, electronic and thermodynamic properties of cubic and orthorhombic XZrS3 ( X = Ba, Sr, Ca ) compounds. J. Comput. Electron. 2019, 2, 415–427. [Google Scholar] [CrossRef]
- Niu, S.; Milam-Guerrero, J.; Melot, B.C. Thermal stability study of transition metal perovskite sulfides. J. Mater. Res. 2018, 24, 4135–4143. [Google Scholar] [CrossRef] [Green Version]
- Perera, S.; Hui, H.; Zhao, C.; Xue, H.; Sun, F.; Deng, C.; Gross, N.; Milleville, C.; Xu, X.; Watson, D.F.; et al. Chalcogenide perovskites – an emerging class of ionic semiconductors. Nano Energy 2016, 22, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Hui, H.; Zhao, C.; Deng, C.; Han, M.; Yu, Z.; Sheng, A.; Roy, P.; Chen, A.; Lin, J.; et al. Realization of BaZrS3 chalcogenide perovskite thin films for optoelectronics. Nano Energy 2020, 68, 104317. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Sun, Q.; Chen, H.; Yin, W.-J. Disparity of the Nature of the Band Gap between Halide and Chalcogenide Single Perovskites for Solar Cell Absorbers. J. Phys. Chem. Lett. 2019, 10, 4566–4570. [Google Scholar] [CrossRef]
- Niu, S.; Huyan, H.; Liu, Y.; Yeung, M.; Ye, K.; Blankemeier, L.; Orvis, T.; Sarkar, D.; Singh, D.J.; Kapadia, R.; et al. Bandgap Control via Structural and Chemical Tuning of Transition Metal Perovskite Chalcogenides. Adv. Matter. 2017, 29, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. J. Non. Cryst. Solids 1995, 1, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total–energy calculations using a plane–wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 18, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 22, 224106. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented–wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT–D) for the 94 elements H–Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pack, J.D.; Monkhorst, H.J. Special points for Brillouin–zone integrations. Phys. Rev. B 1977, 16, 1748–1749. [Google Scholar] [CrossRef]
- Sipe, J.E.; Ghahramani, E. Nonlinear optical response of semiconductors in the independent–particle approximation. Phys. Rev. B 1993, 48, 11705. [Google Scholar] [CrossRef]
- Terentjev, A.V.; Constantin, L.A.; Pitarke, J.M. Gradient–dependent exchange–correlation kernel for materials optical properties. Phys. Rev. B 2018, 98, 085123. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Holec, D.; Mayrhofer, P.H. First–principles study of elastic properties of cubic Cr1–xAlxN alloys. J. Appl. Phys. 2013, 113, 043511. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Zhao, E.; Xiang, H.; Hao, X.; Liu, X.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 76, 1–15. [Google Scholar]
- Lee, C.S.; Kleinke, K.M.; Kleinke, H. Synthesis, structure, and electronic and physical properties of the two SrZrS3 modifications. Solid State Sci. 2005, 7, 1049–1054. [Google Scholar] [CrossRef]
- Lelieveld, R.; IJdo, D.J.W. Sulphides with the GdFeO3 structure. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1980, 36, 2223–2226. [Google Scholar] [CrossRef]
- Niu, S.; Milam–Guerrero, J.; Zhuo, Y.; Ye, K.; Zhao, B.; Melot, B.C.; Ravichandran, J. Thermal stability study of transition metal perovskite sulfides. J. Mater. Res. 2018, 33, 4135–4143. [Google Scholar] [CrossRef] [Green Version]
- Edrees, S.J.; Shukur, M.M.; Obeid, M.M. Computational Condensed Matter First–principle analysis of the structural, mechanical, optical and electronic properties of wollastonite monoclinic polymorph. Comput. Condens. Matter. 2018, 14, 20–26. [Google Scholar] [CrossRef]
- Dzade, N.Y.; De Leeuw, N.H. Periodic DFT+U investigation of the bulk and surface properties of marcasite (FeS2). Phys. Chem. Chem. Phys. 2017, 19, 27478–27488. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B. Condens. Matter Mater. Phys. 2014, 90, 224104. [Google Scholar] [CrossRef] [Green Version]
- Birch, F. Finite Elastic Strain of Cubic Crystals. Phys. Rev. 1947, 71, 809. [Google Scholar] [CrossRef]
- Murnaghan, F.D. The Compressibility of Media under Extreme Pressures. Proc. Natl. Acad. Sci. USA 1944, 30, 244. [Google Scholar] [CrossRef] [Green Version]
- Angelidi, M.; Vassilopoulos, A.P.; Keller, T. Displacement rate and structural effects on Poisson ratio of a ductile structural adhesive in tension and compression. Int. J. Adhes. Adhes. 2017, 78, 13–22. [Google Scholar] [CrossRef]
- Rasukkannu, M.; Velauthapillai, D.; Vajeeston, P. Computational Modeling of Novel Bulk Materials for the Intermediate–Band Solar Cells. ACS Omega 2017, 2, 1454–1462. [Google Scholar] [CrossRef] [PubMed]
- Riffe, D.M. Temperature dependence of silicon carrier effective masses with application to femtosecond reflectivity measurements. J. Opt. Soc. Am. B 2002, 19, 1092–1100. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-S.; Hummer, K.; Kresse, G. Accurate Band Structures and Effective Masses for InP, InAs, and InSb Using Hybrid Functionals. Phys. Rev. B 2009, 80, 035203. [Google Scholar] [CrossRef]
- Bahers, T.L.; Rѐrat, M.; Sautet, P. Semiconductors Used in Photovoltaic and Photocatalytic Devices: Assessing Fundamental Properties from DFT. J. Phys. Chem. C 2014, 118, 5997–6008. [Google Scholar] [CrossRef]
- Dong, M.; Zhang, J.; Yu, J. Effect of Effective Mass and Spontaneous Polarization on Photocatalytic Activity of Wurtzite and Zinc–Blende ZnS. APL Mater. 2015, 3, 104404. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Liu, L.; Zhou, Z. First–Principles Studies on Facet–Dependent Photocatalytic Properties of Bismuth Oxyhalides (BiOXs). RSC Adv. 2012, 2, 9224–9229. [Google Scholar] [CrossRef]
- Faraji, M.; Sabzali, M.; Yousefzadeh, S.; Sarikhani, N.; Ziashahabi, A.; Zirak, M.; Moshfegh, A. Band Engineering and Charge Separation in the Mo1−xWxS2/TiO2 Heterostructure by Alloying: First Principle Prediction. RSC Adv. 2015, 5, 28460–28466. [Google Scholar] [CrossRef]
- Roknuzzaman, M.; Zhang, C.; Ostrikov, K.; Du, A.; Wang, H.; Wang, L.; Tesfamichael, T. Electronic and optical properties of lead–free hybrid double perovskites for photovoltaic and optoelectronic applications. Sci. Rep. 2019, 9, 718. [Google Scholar] [CrossRef] [Green Version]
- Primak, W. Refractive Index of Silicon. Appl. Opt. 1971, 10, 759–763. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| α–SrZrS3 | β–SrZrS3 | |||||
|---|---|---|---|---|---|---|
| Parameter | PBE–D3 | HSE06+D3 | Experiment [32] | PBE+D3 | HSE06+D3 | Experiment [32] |
| a (Å) | 8.551 | 8.540 | 8.525 | 7.133 | 7.125 | 7.109 |
| b (Å) | 3.814 | 3.810 | 3.826 | 9.783 | 9.772 | 9.766 |
| c (Å) | 13.930 | 13.908 | 13.925 | 6.752 | 6.748 | 6.735 |
| d(Sr–S) | 3.06–3.09 | 3.05–3.09 | 3.06–3.08 | 2.99–3.16 | 2.98–3.21 | 2.96–3.37 |
| d(Zr–S) | 2.45–2.65 | 2.44–2.63 | 2.44–2.64 | 2.54–2.57 | 2.54–2.56 | 2.53–2.56 |
| Parameter | α–SrZrS3 | β–SrZrS3 |
|---|---|---|
| C11 | 137.3 | 50.3 |
| C22 | 107.4 | 242.3 |
| C33 | 74.3 | 118.5 |
| C44 | 32.0 | 50.9 |
| C55 | 35.4 | 29.3 |
| C66 | 56.3 | 8.9 |
| C12 | 44.4 | 53.1 |
| C13 | 63.6 | 34.9 |
| C23 | 10.3 | 66.0 |
| B (GPa) | 61.73 | 79.9 |
| G (GPa) | 38.12 | 34.96 |
| E (GPa) | 95.36 | 91.53 |
| υ | 0.244 | 0.436 |
| Material | Direction | m∗h (me) | m∗e (me) | D= m∗h/m∗e |
|---|---|---|---|---|
| α–SrZrS3 | Γ−X | 0.011 | 0.004 | 2.75 |
| X−S | 0.010 | 0.007 | 1.43 | |
| S−Y | 0.015 | 0.013 | 1.15 | |
| Y− Γ | 0.080 | 0.003 | 26.67 | |
| Γ−Z | 0.007 | 0.005 | 1.40 | |
| Z−U | 0.020 | 0.004 | 5.00 | |
| U−R | 0.016 | 0.005 | 3.20 | |
| R−T | 0.023 | 0.049 | 0.47 | |
| T− Γ | 0.021 | 0.004 | 5.25 | |
| β–SrZrS3 | Γ−X | 0.054 | 0.004 | 13.50 |
| X−S | 0.014 | 0.003 | 4.67 | |
| S−Y | 0.017 | 0.067 | 0.25 | |
| Y− Γ | 0.026 | 0.011 | 2.36 | |
| Γ−Z | 0.017 | 0.005 | 3.40 | |
| Z−U | 0.037 | 0.005 | 7.40 | |
| U−R | 0.007 | 0.003 | 2.33 | |
| R−T | 0.012 | 0.015 | 0.80 | |
| T− Γ | 0.009 | 0.008 | 1.13 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eya, H.I.; Ntsoenzok, E.; Dzade, N.Y. First–Principles Investigation of the Structural, Elastic, Electronic, and Optical Properties of α– and β–SrZrS3: Implications for Photovoltaic Applications. Materials 2020, 13, 978. https://doi.org/10.3390/ma13040978
Eya HI, Ntsoenzok E, Dzade NY. First–Principles Investigation of the Structural, Elastic, Electronic, and Optical Properties of α– and β–SrZrS3: Implications for Photovoltaic Applications. Materials. 2020; 13(4):978. https://doi.org/10.3390/ma13040978
Chicago/Turabian StyleEya, Henry Igwebuike, Esidor Ntsoenzok, and Nelson Y. Dzade. 2020. "First–Principles Investigation of the Structural, Elastic, Electronic, and Optical Properties of α– and β–SrZrS3: Implications for Photovoltaic Applications" Materials 13, no. 4: 978. https://doi.org/10.3390/ma13040978