R-Silsesquioxane-Based Network Polymers by Fluoride Catalyzed Synthesis: An Investigation of Cross-Linker Structure and Its Influence on Porosity


Abstract
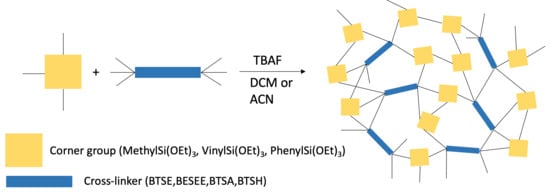
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
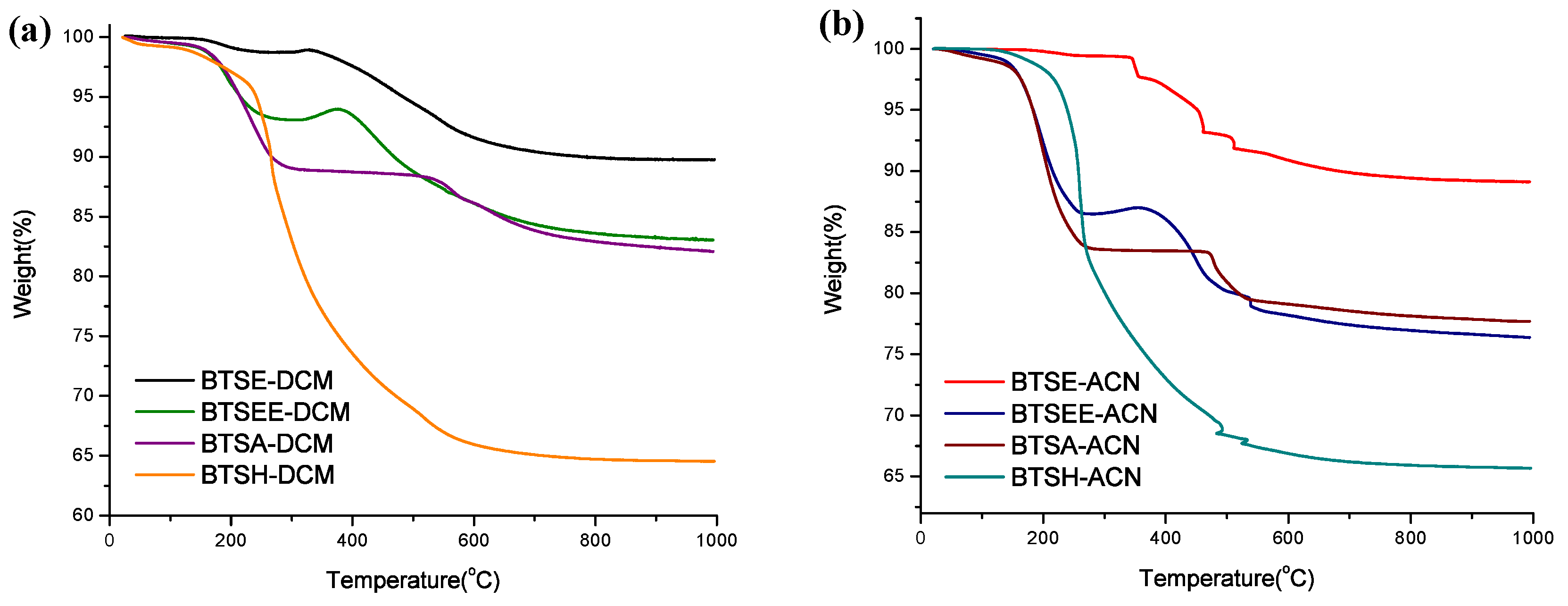
3.1. Cross-Linkers
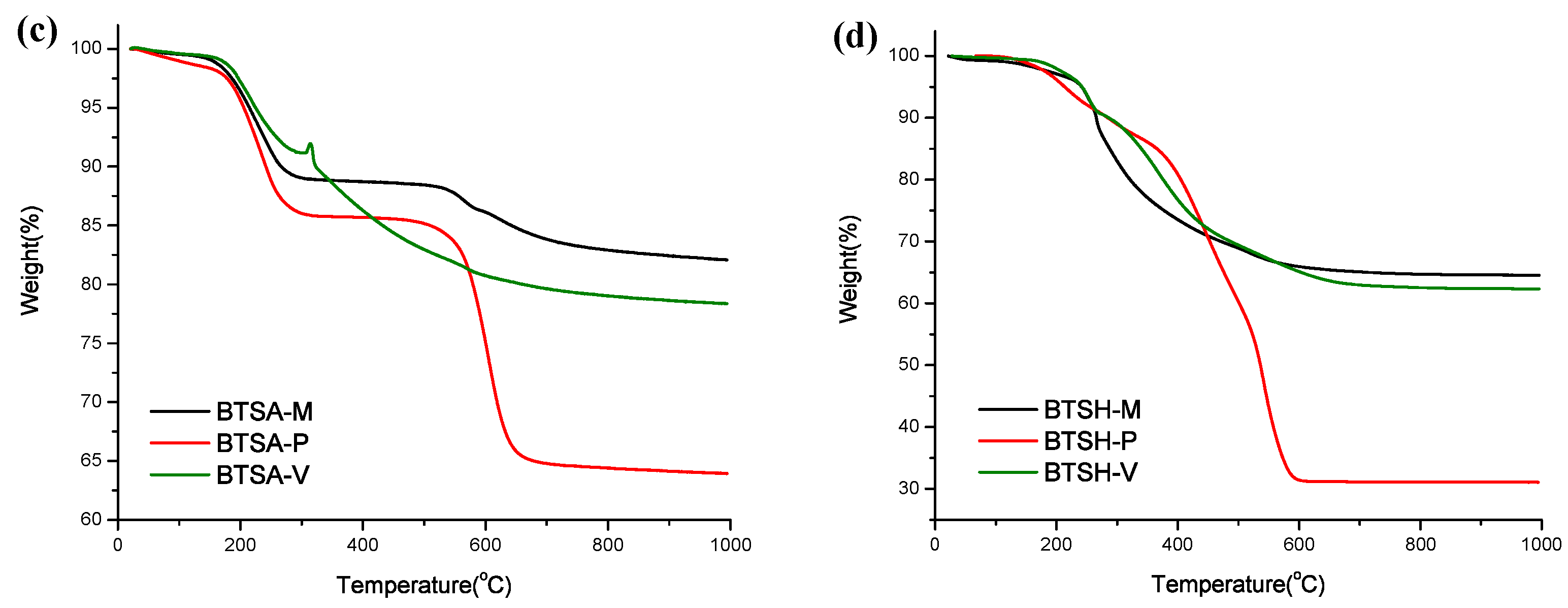
3.2. Corner-Groups
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brinker, C.J.; Scherer, G. Sol-Gel Science, 1st ed.; Academic Press, Inc.: Boston, MA, USA, 1991. [Google Scholar]
- Mizoshita, N.; Tani, T.; Inagaki, S. Syntheses, properties and applications of periodic mesoporous organosilicas prepared from bridged organosilane precursors. Chem. Soc. Rev. 2011, 40, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Loy, D.A.; Shea, K.J. Bridged polysilsesquioxanes. highly porous hybrid organic-inorganic materials. Chem. Rev. 1995, 95, 1431–1442. [Google Scholar] [CrossRef]
- Shea, K.J.; Loy, D.A. A mechanistic investigation of gelation. The sol-gel polymerization of precursors to bridged polysilsesquioxanes. Acc. Chem. Res. 2001, 34, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Scholze, H.; Kaiser, A. Principles of hydrolysis and condensation reaction of alkoxysilanes. J. Non.-Cryst. Solids 1984, 63, 1–11. [Google Scholar] [CrossRef]
- Croissant, J.G.; Cattoën, X.; Durand, J.O.; Chi Man, M.W.; Khashab, N.M. Organosilica hybrid nanomaterials with a high organic content: Syntheses and applications of silsesquioxanes. Nanoscale 2016, 8, 19945–19972. [Google Scholar] [CrossRef]
- Li, Q.; Afeworki, M.; Callen, N.M.; Colby, R.J.; Gopinadhan, M.; Kochersperger, M.L.N.; Peterson, B.K.; Sansone, M.; Weston, S.C.; Calabro, D.C. Template-free self-assembly of mesoporous organosilicas. Chem. Mater. 2018, 30, 2218–2228. [Google Scholar] [CrossRef]
- Barczak, M.; McDonagh, C.; Wencel, D. Micro- and nanostructured sol-gel-based materials for optical chemical sensing (2005–2015). Microchim. Acta 2016, 183, 2085–2109. [Google Scholar] [CrossRef]
- Moreau, J.J.E.; Pichon, B.P.; Chi Man, M.W.; Bied, C.; Pritzkow, H.; Bantignies, J.L.; Dieudonné, P.; Sauvajol, J.L. A better understanding of the self-structuration of bridged silsesquioxanes. Angew. Chem. Int. Ed. 2003, 43, 203–206. [Google Scholar] [CrossRef]
- Díaz-Morales, U.; Bellussi, G.; Carati, A.; Millini, R.; Parker, W.O.N.; Rizzo, C. Ethane-silica hybrid material with ordered hexagonal mesoporous structure. Microporous Mesoporous Mater. 2006, 87, 185–191. [Google Scholar] [CrossRef]
- Boury, B.; Corriu, R.J.P.; Delord, P.; Le Strat, V. Structure of silica-based organic-inorganic hybrid xerogel. J. Non.-Cryst. Solids 2000, 265, 41–50. [Google Scholar] [CrossRef]
- Moreau, J.J.E.; Vellutini, L.; Chi Man, M.W.; Bied, C. Shape-controlled bridged silsesquioxanes: Hollow tubes and spheres. Chem. A Eur. J. 2003, 9, 1594–1599. [Google Scholar] [CrossRef] [PubMed]
- Corriu, R.J.P. The control of nanostructured solids: A challenge for molecular chemistry. Eur. J. Inorg. Chem. 2001, 1109–1121. [Google Scholar] [CrossRef]
- Pons, A.; Casas, L.; Estop, E.; Molins, E.; Harris, K.D.M.; Xu, M. A new route to aerogels: Monolithic silica cryogels. J. Non.-Cryst. Solids 2012, 358, 461–469. [Google Scholar] [CrossRef]
- Boury, B.; Corriu, R.J.P.; Le Strat, V.; Delord, P. Generation of porosity in a hybrid organic-inorganic xerogel by chemical treatment. New J. Chem. 1999, 23, 531–538. [Google Scholar] [CrossRef]
- Barton, T.J.; Bull, L.M.; Klemperer, W.G.; Loy, D.A.; McEnaney, B.; Misono, M.; Monson, P.A.; Pez, G.; Schere, G.W.; Vartuli, J.C.; et al. Tailored porous materials. Chem. Mater. 1999, 11, 2633–2656. [Google Scholar] [CrossRef] [Green Version]
- Ge, M.; Liu, H. Fluorine-containing silsesquioxane-based hybrid porous polymers mediated by bases and their use in water remediation. Chem. A Eur. J. 2018, 24, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, K. Monolithic silsesquioxane materials with well-defined pore structure. J. Mater. Res. 2014, 29, 2773–2786. [Google Scholar] [CrossRef]
- Wang, S.; Tan, L.; Zhang, C.; Hussain, I.; Tan, B. Novel POSS-based organic–inorganic hybrid porous materials by low cost strategies. J. Mater. Chem. A 2015, 3, 6542–6548. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, D.; Li, L.; Yang, W.; Feng, S.; Liu, H. Hybrid porous polymers constructed from octavinylsilsesquioxane and benzene via Friedel-Crafts reaction: Tunable porosity, gas sorption, and postfunctionalization. J. Mater. Chem. A 2014, 2, 2160. [Google Scholar] [CrossRef]
- Chaikittisilp, W.; Kubo, M.; Moteki, T.; Sugawara-Narutaki, A.; Shimojima, A.; Okubo, T. Porous siloxane-organic hybrid with ultrahigh surface area through simultaneous polymerization-destruction of functionalized cubic siloxane cages. J. Am. Chem. Soc. 2011, 133, 13832–13835. [Google Scholar] [CrossRef]
- Cerveau, G.; Corriu, R.J.P.; Framery, E. Influence of the nature of the catalyst on the textural properties of organosilsesquioxane materials. Polyhedron 2000, 19, 307–313. [Google Scholar] [CrossRef]
- Díaz, U.; García, T.; Velty, A.; Corma, A. Hybrid organic-inorganic catalytic porous materials synthesized at neutral pH in absence of structural directing agents. J. Mater. Chem. 2009, 19, 5970–5979. [Google Scholar] [CrossRef]
- Burkett, C.M.; Edmiston, P.L. Highly swellable sol-gels prepared by chemical modification of silanol groups prior to drying. J. Non.-Cryst. Solids 2005, 351, 3174–3178. [Google Scholar] [CrossRef]
- Burkett, C.M.; Underwood, L.A.; Volzer, R.S.; Baughman, J.A.; Edmiston, P.L. Organic-inorganic hybrid materials that rapidly swell in non-polar liquids: Nanoscale morphology and swelling mechanism. Chem. Mater. 2008, 20, 1312–1321. [Google Scholar] [CrossRef]
- Shea, K.J.; Loy, D.A. Bridged polysilsesquioxanes. Molecular-engineered hybrid organic-inorganic materials. Chem. Mater. 2001, 13, 3306–3319. [Google Scholar] [CrossRef]
- Furgal, J.C.; Yamane, H.; Odykirk, T.R.; Yi, E.; Chujo, Y.; Laine, R.M. High surface area, thermally stable, hydrophobic, microporous, rigid gels generated at ambient from MeSi(OEt)3/(EtO)3SiCH2CH2Si(OEt)3 mixtures by F−-catalyzed hydrolysis. Chem. A Eur. J. 2018, 24, 274–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furgal, J.C.; Goodson, T., III; Laine, R.M. D 5h [PhSiO1.5]10 synthesis via F− catalyzed rearrangement of [PhSiO 1.5] n. An experimental/computational analysis of likely reaction pathways. Dalt. Trans. 2016, 45, 1025–1039. [Google Scholar] [CrossRef]
- Hu, N.; Lenora, C.U.; May, T.A.; Hershberger, N.C.; Furgal, J.C. In-situ formed methyl-co-(bis-R) silsesquioxane based polymer networks with solvent controlled pore size distributions and high surface areas. Mater. Chem. Front. 2020, 4, 851–861. [Google Scholar] [CrossRef]
- Kuroda, K.; Shimojima, A.; Kawahara, K.; Wakabayashi, R.; Tamura, Y.; Asakura, Y.; Kitahara, M. Utilization of alkoxysilyl groups for the creation of structurally controlled siloxane-based nanomaterials. Chem. Mater. 2014, 26, 211–220. [Google Scholar] [CrossRef]
- Loy, D.A.; Jamison, G.M.; Baugher, B.M.; Myers, S.A.; Assink, R.A.; Shea, K.J. Sol-gel synthesis of hybrid organic-inorganic materials. Hexylene- and phenylene-bridged polysiloxanes. Chem. Mater. 1996, 8, 656–663. [Google Scholar] [CrossRef]
- Hu, L.C.; Shea, K.J. Organo-silica hybrid functional nanomaterials: How do organic bridging groups and silsesquioxane moieties work hand-in-hand? Chem. Soc. Rev. 2011, 40, 688–695. [Google Scholar] [CrossRef]
- Singh, G.; Kim, I.Y.; Lakhi, K.S.; Srivastava, P.; Naidu, R.; Vinu, A. Single step synthesis of activated bio-carbons with a high surface area and their excellent CO2 adsorption capacity. Carbon 2017, 116, 448–455. [Google Scholar] [CrossRef]
- Long, H.; Harley-Trochimczyk, A.; Pham, T.; Tang, Z.; Shi, T.; Zettl, A.; Carraro, C.; Worsley, M.A.; Maboudian, R. High surface area MoS2/graphene hybrid aerogel for ultrasensitive NO2 detection. Adv. Funct. Mater. 2016, 26, 5158–5165. [Google Scholar] [CrossRef] [Green Version]
- Duan, L.; Zhou, Z.; Yao, B. Fabrication, structural characterization and uniaxial tensile properties of novel sintered multi-layerwire mesh porous plates. Materials 2018, 11, 156. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Ma, X.; Chen, R.; Wang, C.; Lu, M. Nitrogen-containing functional groups-facilitated acetone adsorption by ZIF-8-derived porous carbon. Materials 2018, 11, 159. [Google Scholar] [CrossRef] [Green Version]
- Fertier, L.; Théron, C.; Carcel, C.; Trens, P.; Wong Chi Man, M. PH-responsive bridged silsesquioxane. Chem. Mater. 2011, 23, 2100–2106. [Google Scholar] [CrossRef]
- Randall, J.P.; Meador, M.A.B.; Jana, S.C. Polymer reinforced silica aerogels: Effects of dimethyldiethoxysilane and bis(trimethoxysilylpropyl)amine as silane precursors. J. Mater. Chem. A 2013, 1, 6642–6652. [Google Scholar] [CrossRef]
- Guo, S.; Okubo, T.; Kuroda, K.; Shimojima, A. A photoresponsive azobenzene-bridged cubic silsesquioxane network. J. Sol-Gel Sci. Technol. 2016, 79, 262–269. [Google Scholar] [CrossRef]
- Radi, B.; Wellard, R.M.; George, G.A. Controlled poly(ethylene glycol) network structures through silsesquioxane cross-links formed by sol-gel reactions. Macromolecules 2010, 43, 9957–9963. [Google Scholar] [CrossRef]
- Yen, Y.C.; Ye, Y.S.; Cheng, C.C.; Lu, C.H.; Tsai, L.D.; Huang, J.M.; Chang, F.C. The effect of sulfonic acid groups within a polyhedral oligomeric silsesquioxane containing cross-linked proton exchange membrane. Polymer 2010, 51, 84–91. [Google Scholar] [CrossRef]
- Jeon, J.H.; Lim, J.H.; Kim, K.M. Organic-inorganic hybrid nanocomposites of poly(sodium 4-styrenesulfonate) and octafunctional polyhedral oligomeric silsesquioxane (POSS). Macromol. Res. 2010, 18, 341–345. [Google Scholar] [CrossRef]
- Li, Y.; Dong, X.-H.; Zou, Y.; Wang, Z.; Yue, K.; Huang, M.; Liu, H.; Feng, X.; Lin, Z.; Zhang, W.; et al. Polyhedral oligomeric silsesquioxane meets “click” chemistry: Rational design and facile preparation of functional hybrid materials. Polymer 2017, 125, 303–329. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Sangtrirutnugul, P.; Chaiprasert, T.; Hunsiri, W.; Jitjaroendee, T.; Songkhum, P.; Laohhasurayotin, K.; Osotchan, T.; Ervithayasuporn, V. Tunable porosity of cross-linked-polyhedral oligomeric silsesquioxane supports for palladium-catalyzed aerobic alcohol oxidation in water. ACS Appl. Mater. Interfaces 2017, 9, 12812–12822. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Matsukawa, K.; Miyata, T.; Okubo, T.; Kuroda, K.; Shimojima, A. Photoinduced bending of self-assembled azobenzene-siloxane hybrid. J. Am. Chem. Soc. 2015, 137, 15434–15440. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Gan, Q. Synthesis and luminescence properties of spherical bridged polysilsequioxanes activated by lanthanide ions. J. Sol-Gel Sci. Technol. 2010, 56, 141–144. [Google Scholar] [CrossRef]
- Bürglová, K.; Noureddine, A.; Hodačová, J.; Toquer, G.; Cattoën, X.; Wongchiman, M. A general method for preparing bridged organosilanes with pendant functional groups and functional mesoporous organosilicas. Chem. A Eur. J. 2014, 20, 10371–10382. [Google Scholar] [CrossRef]
- Cornelius, M.; Hoffmann, F.; Ufer, B.; Behrens, P.; Fröba, M. Systematic extension of the length of the organic conjugated π-system of mesoporous silica-based organic-inorganic hybrid materials. J. Mater. Chem. 2008, 18, 2587–2592. [Google Scholar] [CrossRef]
- Dirè, S.; Tagliazucca, V.; Callone, E.; Quaranta, A. Effect of functional groups on condensation and properties of sol-gel silica nanoparticles prepared by direct synthesis from organoalkoxysilanes. Mater. Chem. Phys. 2011, 126, 909–917. [Google Scholar] [CrossRef]
- Blanco, I. The rediscovery of POSS: A molecule rather than a filler. Polymers 2018, 10, 904. [Google Scholar] [CrossRef] [Green Version]
- Pan, D.; Yi, E.; Doan, P.H.; Furgal, J.C.; Schwartz, M.; Clark, S.; Goodson, T., III; Laine, R.M. Microporous inorganic/organic hybrids via oxysilylation of a cubic symmetry nanobuilding block [(HMe2SiOSiO1.5)8] with RxSi(OEt)4−x. J. Ceram. Soc. Jpn. 2015, 123, 756–763. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yin, G.; Li, Z.; Wu, P.; Jin, X.; Li, Q. The preparation and chemical structure analysis of novel POSS-based porous materials. Materials 2019, 12, 1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Yu, J.; Xue, L.; Zhang, C.; Zha, Y.; Gu, Y. Investigation of molecular structure and thermal properties of thermo-oxidative aged SBS in blends and their relations. Materials 2017, 10, 768. [Google Scholar] [CrossRef]
- Bautista, Y.; Gozalbo, A.; Mestre, S.; Sanz, V. Thermal degradation mechanism of a thermostable polyester stabilized with an open-cage oligomeric silsesquioxane. Materials 2017, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Loy, D.A.; Jamison, G.M.; Baugher, B.M.; Russick, E.M.; Assink, R.A.; Prabakar, S.; Shea, K.J. Alkylene-bridged polysilsesquioxane aerogels: Highly porous hybrid organic-inorganic materials. J. Non.-Cryst. Solids 1995, 186, 44–53. [Google Scholar] [CrossRef]
- Feinle, A.; Elsaesser, M.S.; Hüsing, N. Sol-gel synthesis of monolithic materials with hierarchical porosity. Chem. Soc. Rev. 2016, 45, 3377–3399. [Google Scholar] [CrossRef]
- Chen, T.; Li, M.; Liu, J. π-π Stacking Interaction: A nondestructive and facile means in material engineering for bioapplications. Cryst. Growth Des. 2018, 18, 2765–2783. [Google Scholar] [CrossRef]
- Ronchi, M.; Sulaiman, S.; Boston, N.R.; Laine, R.M. Fluoride catalyzed rearrangements of polysilsesquioxanes, mixed Me, vinyl T8, Me, vinyl T10 and T12 cages. Appl. Organomet. Chem. 2009, 24, 551–557. [Google Scholar] [CrossRef] [Green Version]
- Krug, D.J.; Asuncion, M.Z.; Laine, R.M. Facile Approach to Recycling Highly Cross-Linked Thermoset Silicone Resins under Ambient Conditions. ACS Omega 2019, 4, 3782–3789. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cross-Linker | Solvent | P = Particle N = No Gel | Surface Area (m2 g−1) | Temperature at 5% Mass Loss (T5%) | Residue at 1000 °C (Ceramic Yield) |
|---|---|---|---|---|---|
| BTSE | |||||
| DCM | P | 1076 | 361.3 °C | 86.2% | |
| ACN | P | 1022 | 451.6 °C | 89.2% | |
| BTSEE | |||||
| DCM | P | 556 | 251.2 °C | 83.1% | |
| ACN | P | 423 | 183.4 °C | 76.5% | |
| BTSA | |||||
| DCM | P | 454 | 214.7 °C | 82.2% | |
| ACN | P | 212 | 182.9 °C | 77.7% | |
| BTSH | |||||
| DCM | N | 112 | 241.4 °C | 64.6% | |
| ACN | N | 414 | 239.2 °C | 65.7% |
| Cross-Linker | Corner Silane | P = Particle N = No Gel | Surface Area (m2 g−1) | Temperature at 5% Mass Loss (T5%) | Residue at 1000 °C (Ceramic Yield) |
|---|---|---|---|---|---|
| BTSE | |||||
| MeSi(OEt)3 (methyl) | P | 1076 | 483.7 °C | 89.7% | |
| PhSi(OEt)3 (phenyl) | P | 1 | 278.9 °C | 54.6% | |
| VinylSi(OEt)3 (vinyl) | P | 553 | 315.2 °C | 77.5% | |
| BTSEE | |||||
| MeSi(OEt)3 | P | 556 | 229.1 °C | 83.1% | |
| PhSi(OEt)3 | P | 245 | 216.8 °C | 57.8% | |
| VinylSi(OEt)3 | P | 455 | 369.3 °C | 78.0% | |
| BTSA | |||||
| MeSi(OEt)3 | P | 454 | 214.7 °C | 82.2% | |
| PhSi(OEt)3 | P | 316 | 205.7 °C | 64.0% | |
| VinylSi(OEt)3 | P | 492 | 225.0 °C | 78.5% | |
| BTSH | |||||
| MeSi(OEt)3 | N | 112 | 241.4 °C | 64.6% | |
| PhSi(OEt)3 | N | - | 213.5 °C | 31.1% | |
| VinylSi(OEt)3 | N | - | 242.0 °C | 62.4% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, N.-h.; Furgal, J.C. R-Silsesquioxane-Based Network Polymers by Fluoride Catalyzed Synthesis: An Investigation of Cross-Linker Structure and Its Influence on Porosity. Materials 2020, 13, 1849. https://doi.org/10.3390/ma13081849
Hu N-h, Furgal JC. R-Silsesquioxane-Based Network Polymers by Fluoride Catalyzed Synthesis: An Investigation of Cross-Linker Structure and Its Influence on Porosity. Materials. 2020; 13(8):1849. https://doi.org/10.3390/ma13081849
Chicago/Turabian StyleHu, Nai-hsuan, and Joseph C. Furgal. 2020. "R-Silsesquioxane-Based Network Polymers by Fluoride Catalyzed Synthesis: An Investigation of Cross-Linker Structure and Its Influence on Porosity" Materials 13, no. 8: 1849. https://doi.org/10.3390/ma13081849