Quantum-Chemical Design of Molecular Structures of Tetra-, Penta- and Hexanuclear Metal Clusters Containing Aluminum and 3d-Element Atoms



Abstract
:1. Introduction
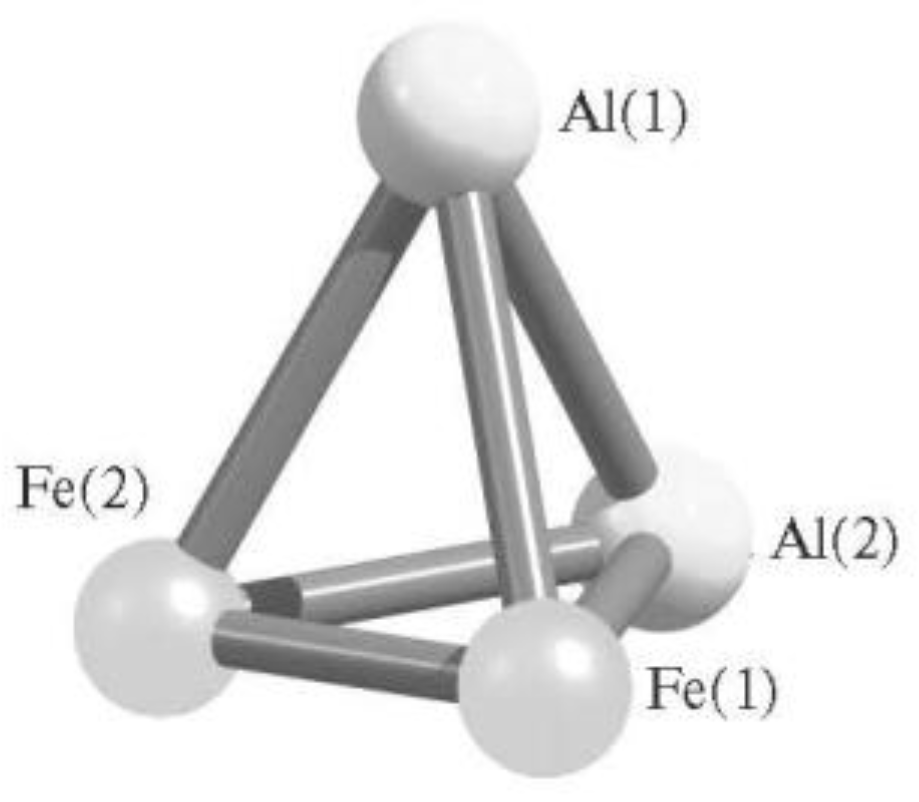
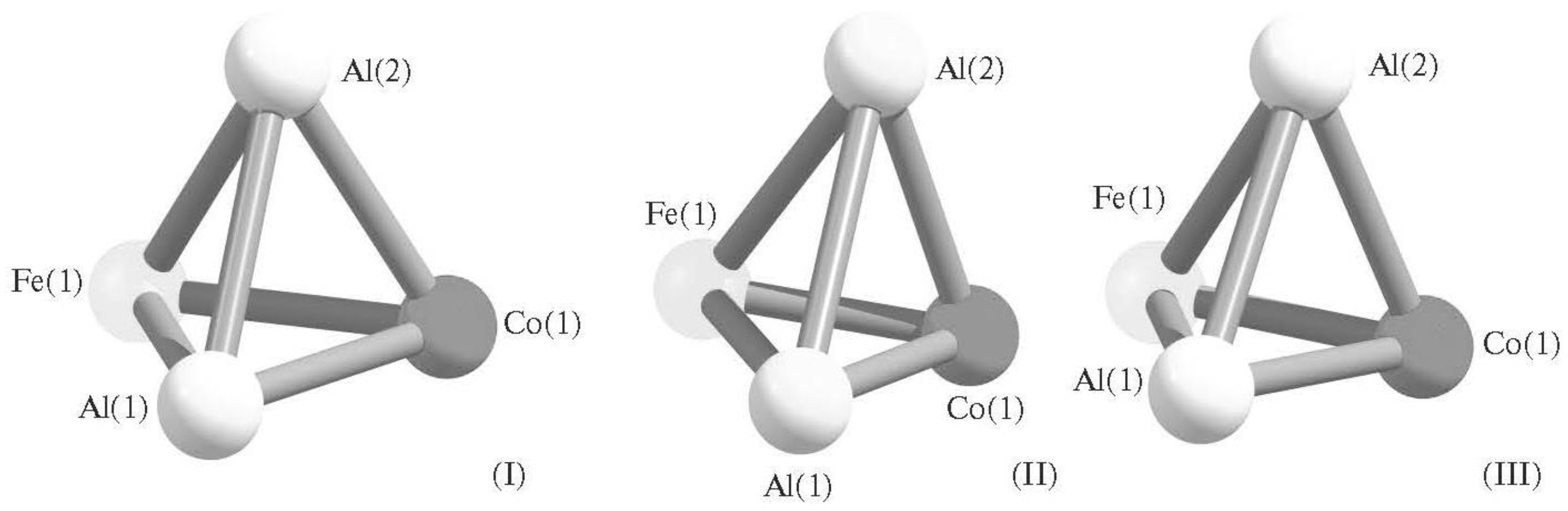
2. Tetranuclear (AlM) Metal Clusters
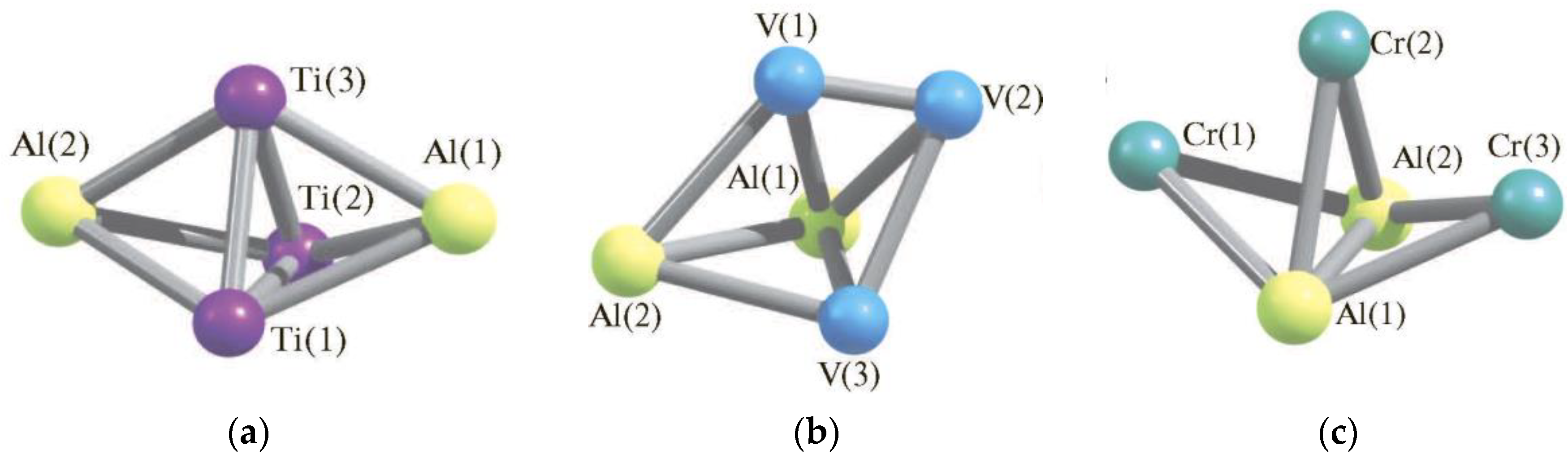
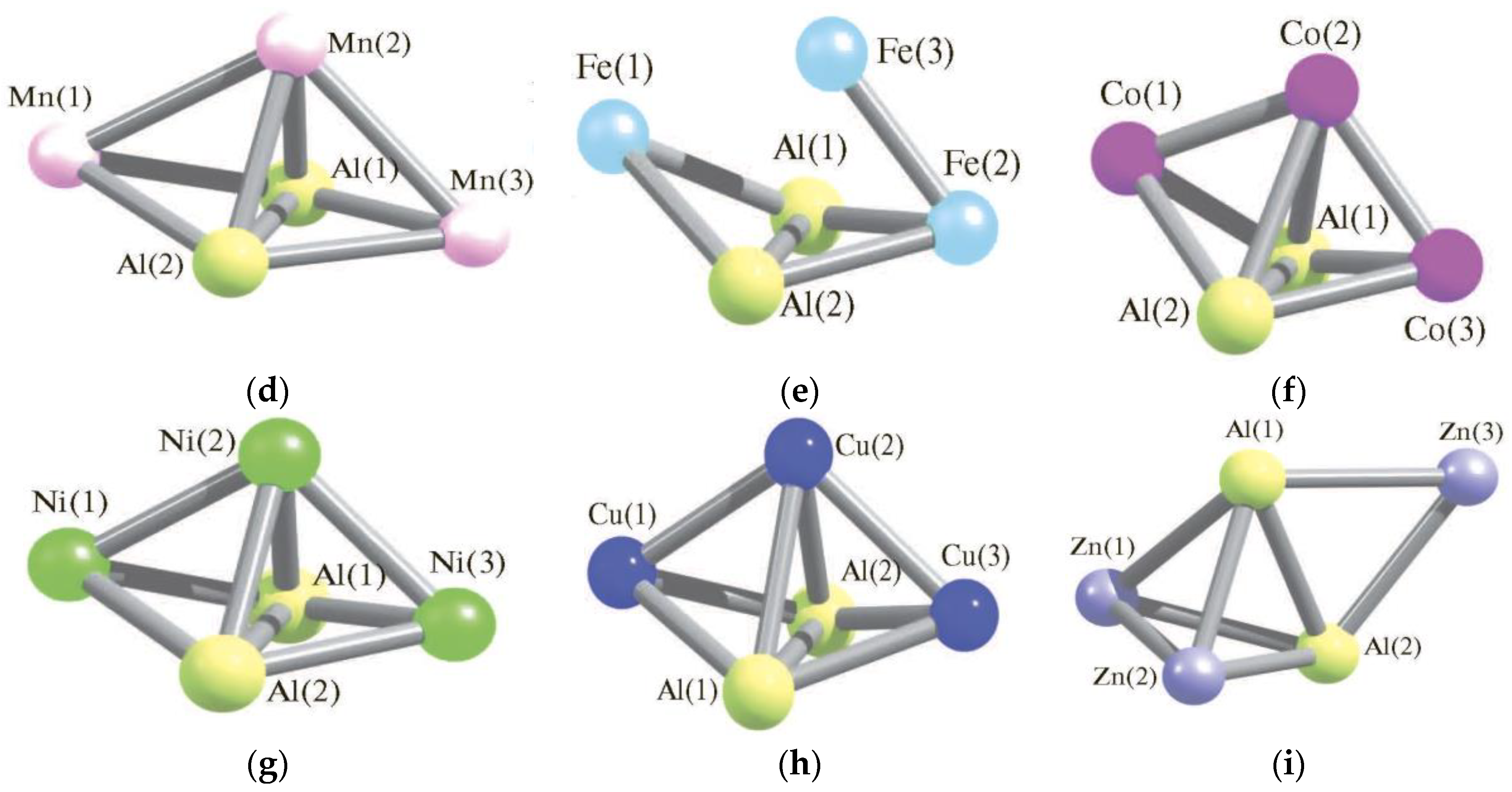
3. Pentanuclear (AlM) Metal Clusters
4. Hexanuclear (AlM) Metal Clusters
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maroun, F.; Ozanam, F.; Magnussen, O.M.; Behm, R.J. The role of atomic ensembles in the reactivity of bimetallic electrocatalysts. Science 2001, 293, 1811–1814. [Google Scholar] [CrossRef] [PubMed]
- Derosa, P.A.; Seminario, J.M.; Balbuena, P.B. Properties of Small Bimetallic Ni−Cu Clusters. J. Phys. Chem. A 2001, 105, 7917–7925. [Google Scholar] [CrossRef]
- Eberhardt, W. Clusters as new materials. Surf. Sci. 2002, 500, 242–270. [Google Scholar] [CrossRef]
- Lu, Q.L.; Zhu, L.Z.; Ma, L.; Wang, G.H. Magnetic properties of Co/Cu and Co/Pt bimetallic clusters. Chem. Phys. Lett. 2005, 407, 176–179. [Google Scholar] [CrossRef]
- Yuan, D.W.; Wang, Y.; Zeng, Z. Geometric, electronic, and bonding properties of Au[sub N]M (N=1–7, M=Ni, Pd, Pt) clusters. J. Chem. Phys. 2005, 122, 114310. [Google Scholar] [CrossRef]
- Wang, X.; Cao, Z.; Lu, X.; Lin, M.; Zhang, Q. Structure and stability of binary transition-metal clusters (NbCo)[sub n] (n ≤ 5): A relativistic density-functional study. J. Chem. Phys. 2005, 123, 064315. [Google Scholar] [CrossRef] [Green Version]
- Barcaro, G.; Fortunelli, A.; Rossi, G.; Nita, F.; Ferrando, R. Electronic and Structural Shell Closure in AgCu and AuCu Nanoclusters. J. Phys. Chem. B 2006, 110, 23197–23203. [Google Scholar] [CrossRef]
- Néel, N.; Kröger, J.; Berndt, R.; Wehling, T.O.; Lichtenstein, A.I.; Katsnelson, M.I. Controlling the kondo effectin CoCun clusters atom by atom. Phys. Rev. Lett. 2008, 101, 266803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, D.; Xiao-Yu, K.; Jian-Jun, G.; Ben-Xia, Z. First-principle study of AunFe (n=1–7) clusters. J. Mol. Struct. 2009, 902, 54–58. [Google Scholar]
- Du, J.; Shen, N.; Zhu, L.; Wang, J. Emergence of noncollinear magnetic ordering in bimetallic Co6− n Mnn clusters. J. Phys. D: Appl. Phys. 2010, 43, 015006. [Google Scholar] [CrossRef]
- Lv, J.; Zhang, F.Q.; Jia, J.F.; Xu, X.H.; Wu, H.S. First-principles study of structural, electronic and magnetic properties of Co13−nMn (n=1, 2, M=Mn, V and Al) clusters. J. Mol. Struct. (TheoChem) 2010, 955, 14–22. [Google Scholar] [CrossRef]
- Kilimis, D.A.; Papageorgiou, D.G. Density functional study of small bimetallic Ag–Pd clusters. J. Mol. Struct. 2010, 939, 112–117. [Google Scholar] [CrossRef]
- Lin, L.; Claes, P.; Gruene, P.; Meijer, G.; Fielicke, A.; Nguyen, M.T.; Lievens, P. Far-Infrared Spectra of Yttrium-Doped Gold Clusters AunY (n = 1-9). ChemPhysChem 2010, 11, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Garbounis, D.N.; Tsipis, A.C.; Tsipis, C.A. Structural, electronic, bonding, magnetic, and optical properties of bimetallic [RunAum]0/+ (n+m ≤ 3) clusters. J. Comput. Chem. 2010, 31, 2836–2852. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Fa, W.; Dong, I. Magnetic Properties of Transition-Metal-Doped Tubular Gold Clusters: M@Au24 (M = V, Cr, Mn, Fe, Co, and Ni). J. Phys. Chem. A 2010, 114, 4031–4035. [Google Scholar] [CrossRef] [PubMed]
- Venkataramanan, R.S.; Sahara, R.; Mizuseki, H.; Kawazoe, Y. Titanium-Doped Nickel Clusters TiNin (n = 1−12): Geometry, Electronic, Magnetic, and Hydrogen Adsorption Properties. J. Phys. Chem. A 2010, 114, 5049–5057. [Google Scholar] [CrossRef]
- Wang, H.Q.; Kuang, X.Y.; Li, H.F. Density functional study of structural and electronic properties of bimetallic copper-gold clusters: Comparison with pure and doped gold clusters. Phys. Chem. Chem. Phys. 2010, 12, 5156. [Google Scholar] [CrossRef]
- Montejano-Carrizales, J.M.; Aguilera-Granja, F.; Morán-López, J.L. Structural and magnetic properties of FemYn (m+n = 7, Y = Ru, Rh, Pd, and Pt) nanoalloys. Eur. Phys. J. D 2011, 64, 53–62. [Google Scholar] [CrossRef]
- Jin, X.X.; Du, J.G.; Jiang, G.; Luo, X.; Wang, X.W. Geometries and electronic properties of NbnV(0, ±1) (n = 1−6) clusters studied by density-functional theory. Eur. Phys. J. D 2011, 64, 323–329. [Google Scholar] [CrossRef]
- Yang, J.X.; Guo, J.J.; Die, D. Ab initio study of AunIr (n=1–8) clusters. Comput. Theor. Chem. 2011, 963, 435–438. [Google Scholar] [CrossRef]
- Zhao, S.; Ren, Y.; Wang, J.; Yin, W. Density functional study of NO binding on small AgnPdm (n+m≤5) clusters. Comput. Theor. Chem. 2011, 964, 298–303. [Google Scholar] [CrossRef]
- Ma, Q.M.; Xie, Z.; Wang, B.R.; Liu, Y.; You-Cheng Li, Y.C. Structure, stability and magnetic moments of the FenCr clusters: All-electron density functional theory investigations. Solid State Commun. 2011, 151, 806–810. [Google Scholar] [CrossRef]
- Aguilera-Granja, F.; Longo, R.C.; Gallego, L.J.; Vega, A. Magnetic Cooperative Effects in Small Ni–Ru Clusters. J. Phys. Chem. A 2011, 115, 13950–13955. [Google Scholar] [CrossRef] [PubMed]
- Nhatab, P.V.; Nguyen, M.T. Trends in structural, electronic and energetic properties of bimetallic vanadium–gold clusters AunV with n = 1-14. Phys. Chem. Chem. Phys. 2011, 13, 16254–16264. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Bai, X.; Jia, J.F.; Xu, X.H.; Wu, H.S. Structural, electronic and magnetic properties of ConRh (n=1–8) clusters from density functional calculations. Phys. B Condens. Matt. 2012, 407, 14–21. [Google Scholar] [CrossRef]
- Hong, L.; Wang, H.; Cheng, J.; Huang, X.; Sai, L.; Zhao, J. Atomic structures and electronic properties of small Au–Ag binary clusters: Effects of size and composition. Comput. Theor. Chem. 2012, 993, 36–44. [Google Scholar] [CrossRef]
- Shewale, V.; Deshpande, M. Structural, electronic, and magnetic properties of NinM clusters (M=Hf, Ta, W) with n=1–12. Comput. Theor. Chem. 2012, 984, 128–136. [Google Scholar] [CrossRef]
- Ding, L.P.; Kuang, X.Y.; Shao, P.; Zhao, Y.R.; Li, Y.F. A comparative study on geometries, stabilities, and electronic properties between bimetallic AgnX (X = Au, Cu; n=1−8) and pure silver clusters. Chin. Phys. B 2012, 21, 043601. [Google Scholar] [CrossRef]
- Yao, J.; Bin Xu, B.; Wang, Y. Ground State Structures, Electronic and Magnetic Properties of ScnFe (n=2-10) Clusters. Chin. J. Chem. 2012, 30, 905–913. [Google Scholar] [CrossRef]
- Florez, E.; Mondragon, F.; Illas, F. Theoretical study of the structure and reactivity descriptors of CunM (M = Ni, Pd, Pt, n = 1–4) bimetallic nanoparticles supported on MgO(001). Surf. Sci. 2012, 606, 1010–1018. [Google Scholar] [CrossRef]
- Ju, W.; Yang, Z. Influence of spin–orbit coupling on electronic structures of TM@Au12 (TM=3d, 4d, and 5d atoms). Phys. Lett. A 2012, 376, 1300–1305. [Google Scholar] [CrossRef]
- Ma, L.; Wang, J.; Hao, Y.; Wang, G. Density functional theory study of FePdn (n=2–14) clusters and interactions with small molecules. Comput. Mater. Sci. 2013, 68, 166–173. [Google Scholar] [CrossRef]
- Liu, X.; Tian, D.; Meng, C. DFT study on stability and H2 adsorption activity of bimetallic Au79−nPdn (n=1–55) clusters. Chem. Phys. 2013, 415, 179–185. [Google Scholar] [CrossRef]
- Tafoughalt, M.A.; Samah, M. Structural properties and relative stability of silver-doped gold clusters AgAun-1 (n = 3–13): Density functional calculations. Comput. Theor. Chem. 2014, 1033, 23–30. [Google Scholar] [CrossRef]
- Wen, J.Q.; Xia, T.; Zhou, H.; Wang, J.F. A density functional theory study of small bimetallic PdnAl (n=1–8) clusters. J. Phys. Chem. Solids 2014, 75, 528–534. [Google Scholar] [CrossRef]
- Singh, N.B.; Sarkar, U. A density functional study of chemical, magnetic and thermodynamic properties of small palladium clusters. Mol. Simul. 2014, 40, 1255–1264. [Google Scholar] [CrossRef]
- Bouderbala, W.; Boudjahem, A.G.; Soltani, A. Geometries, stabilities, electronic and magnetic properties of small PdnIr (n = 1–8) clusters from first principles calculations. Mol. Phys. 2014, 112, 1789–1798. [Google Scholar] [CrossRef]
- Ling, W.; Dong, D.; Shi-Jian, W.; Zheng-Quan, Z. Geometrical, electronic, and magnetic properties of CunFe (n=1–12) clusters: A density functional study. J. Phys. Chem. Solids 2015, 76, 10–16. [Google Scholar] [CrossRef]
- Chaves, A.S.; Rondina, G.G.; Piotrowski, M.J.; Da Silva, J.L.F. Structural formation of binary PtCu clusters: A density functional theory investigation. Comput. Mater. Sci. 2015, 98, 278–286. [Google Scholar] [CrossRef]
- Gong, X.; Ju, W.; Li, T.; Feng, Z.; Wang, Y. Spin–orbit Splitting and Magnetism of Icosahedral M@Ag12 Clusters (M = 3d and 4d atoms). J. Clust. Sci. 2015, 26, 759–773. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. Molekularnye struktury poliyadernykh metalloklasterov po dannym raschyota metodom DFT. (Geterobi)tetrayadernyi klaster Fe2Co2. Herald Technol. Univ. 2016, 19, 20–23. [Google Scholar]
- Die, D.; Zheng, B.X.; Zhao, L.Q.; Zhu, Q.W.; Zhao, Z.Q. Insights into the structural, electronic and magnetic properties of V-doped copper clusters: Comparison with pure copper clusters. Sci. Rep. 2016, 6, 31978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Odail, F.; Mazher, J.; Abuelela, A.M. A density functional theory study of structural, electronic and magnetic properties of small PdnAg (n = 1–8) clusters. Comput. Theor. Chem. 2017, 1125, 103–111. [Google Scholar] [CrossRef]
- Deng, M.; Xin, Z.; Yan, X.; Liu, J.; Yu, M. Structural, Electronic, and Magnetic Properties of Bimetallic NimNbn (m+n ≤ 8) Clusters: First Principle Study. J. Supercond. Novel Magn. 2017, 30, 251–260. [Google Scholar] [CrossRef]
- Zhang, G.; Zhai, Z.; Sheng, Y. Structural, electronic and magnetic properties of TinMo (n = 1-7) clusters. Eur. Phys. J. D 2017, 71, 9. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Lv, J. Equilibrium geometries, electronic structure and magnetic properties of ConSn (n = 1–12) clusters from density functional calculations. Mod. Phys. Lett. B 2017, 31, 1750171. [Google Scholar] [CrossRef]
- Datta, S.; Raychaudhuri, A.K.; Saha-Dasgupta, T. First principles study of bimetallic Ni13− nAgn nano-clusters (n = 0–13): Structural, mixing, electronic, and magnetic properties. J. Chem. Phys. 2017, 146, 164301. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Iwasa, T.; Taketsugu, T. Insights into geometries, stabilities, electronic structures, reactivity descriptors, and magnetic properties of bimetallic NimCun-m (m = 1, 2; n = 3-13) clusters: Comparison with pure copper clusters. J. Comput. Chem. 2018, 39, 1878–1889. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Fei, S.; Huang, L.; Forrey, R.C.; Cheng, H. Tuning the Catalytic Activity of PdxNiy (x + y = 6) Bimetallic Clusters for Hydrogen Dissociative Chemisorption and Desorption. ACS Omega 2019, 4, 12498–12504. [Google Scholar] [CrossRef]
- Ranjan, P.; Chakraborty, T. Structure and electronic properties of AunPt (n = 1–8) nanoalloy clusters: The density functional theory study. J. Nanopart. Res. 2020, 22, 11. [Google Scholar] [CrossRef]
- Deshpande, M.D.; Roy, S.; Kanhere, D.G. Equilibrium geometries, electronic structure, and magnetic properties of NinSn clusters (n=1–12). Phys. Rev. B 2007, 76, 195423. [Google Scholar] [CrossRef]
- Chen, X.; Deng, K.; Liu, Y.; Tang, C.; Yuan, Y.; Tan, W.; Wang, X. The geometric, optical, and magnetic properties of the endohedral stannaspherenes M@Sn[sub 12] (M=Ti, V, Cr, Mn, Fe, Co, Ni). J. Chem. Phys. 2008, 129, 094301. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Liu, M.; Zhu, W.; Deng, K. Probing the geometric, optical, and magnetic properties of 3d transition-metal endohedral Ge12M (M=Sc–Ni) clusters. Comput. Theor. Chem. 2011, 969, 56–60. [Google Scholar] [CrossRef]
- Rohrmann, U.; Schäfer, R. Stern-Gerlach Experiments on Fe@Sn12: Magnetic Response of a Jahn–Teller Distorted Endohedrally Doped Molecular Cage Cluster. J. Phys. Chem. C 2015, 119, 10958–10961. [Google Scholar] [CrossRef]
- Shewale, V.H.; Deshpande, M.D.; Kanhere, D.G. Structures, energetics and magnetic properties of (NiSn)n clusters with n = 1–6. Pramana 2009, 73, 699–710. [Google Scholar] [CrossRef]
- Jaiswal, S.; Kumar, V. Growth behavior and electronic structure of neutral and anion ZrGen (n = 1–21) clusters. Comput. Theor. Chem. 2016, 1075, 87–97. [Google Scholar] [CrossRef]
- Sosa-Hernández, E.M.; Montejano-Carrizales, J.M.; Alvarado-Leyva, P.G. Global Minimum Structures, Stability and Electronic Properties of Small NiXSnY (X+Y≤ 5) Bimetallic Clusters; a DFT Study. Eur. Phys. J. D 2016, 70, 208. [Google Scholar] [CrossRef]
- Zhao, G.; Zhang, J.; Jing, Q.; Luo, Y.; Wang, Y. A density functional study of Y[sub n]Al (n=1–14) clusters. J. Chem. Phys. 2007, 127, 234312. [Google Scholar] [CrossRef]
- Tian, F.Y.; Jing, Q.; Wang, Y.X. Structure, stability, and magnetism of ScnAl (n=1–8,12) clusters: Density-functional theory investigations. Phys. Rev. A 2008, 77, 013202. [Google Scholar] [CrossRef]
- Wang, M.; Qiu, G.; Huang, X.; Du, Z.; Li, Y. Study of the size-dependent properties of ScnAl (n = 1–14) clusters by density-functional theory. J. Phys. Condens. Matter. 2009, 21, 046004. [Google Scholar] [CrossRef]
- Du, J.; Sun, X.; Jiang, G. Structures, chemical bonding, magnetisms of small Al-doped zirconium clusters. Phys. Lett. A 2010, 374, 854–860. [Google Scholar] [CrossRef]
- Hua, Y.; Liu, Y.; Jiang, G.; Du, J.; Chen, J. Geometric Transition and Electronic Properties of Titanium-Doped Aluminum Clusters: AlnTi (n = 2–24). J. Phys. Chem. A 2013, 117, 2590–2597. [Google Scholar] [CrossRef] [PubMed]
- Rusina, G.G.; Borisova, S.D.; Chulkov, E.V. Structure and atomic vibrations in bimetallic Ni13 − nAln clusters. JETP Letters 2015, 101, 474–480. [Google Scholar] [CrossRef]
- Schaefer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Hoe, W.M.; Cohen, A.; Handy, N.C. Assessment of a new local exchange functional OPTX. Chem. Phys. Lett. 2001, 341, 319–328. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Paulsen, H.; Duelund, L.; Winkler, H.; Toftlund, H.; Trautwein, A.X. Free Energy of Spin-Crossover Complexes Calculated with Density Functional Methods. Inorg. Chem. 2001, 40, 2201–2203. [Google Scholar] [CrossRef]
- Swart, M.; Groenhof, A.R.; Ehlers, A.W.; Lammertsma, K. Validation of Exchange−Correlation Functionals for Spin States of Iron Complexes. J. Phys. Chem. A 2004, 108, 5479–5483. [Google Scholar] [CrossRef]
- Swart, M.; Ehlers, A.W.; Lammertsma, K. Performance of the OPBE exchange-correlation functional. Mol. Phys. 2004, 102, 2467–2474. [Google Scholar] [CrossRef]
- Swart, M. Metal–ligand bonding in metallocenes: Differentiation between spin state, electrostatic and covalent bonding. Inorg. Chim. Acta 2007, 360, 179–189. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. Molekulyarnye struktury poliyadernykh metalloklasterov po dannym raschyota metodom DFT. (Geterobi)tetrayadernyi klaster AlFe3. Herald Technol. Univ. 2016, 19, 18–21. [Google Scholar]
- Mikhailov, O.V.; Chachkov, D.V. Models of Molecular Structures of Aluminum–Iron Clusters AlFe3, Al2Fe3, and Al2Fe4 according to Quantum-Chemical DFT Calculations. Russ. J. Inorg. Chem. 2017, 62, 336–343. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Modeli molekularnylh struktur alyuminii-zheleznykh klasterov AlFe3, Al2Fe3 i Al2Fe4 po dannym kvantovo-khimicheskogo rascheta metodom DFT. Zh. Neorg. Khim. 2017, 62, 321–329. [Google Scholar]
- Mikhailov, O.V.; Chachkov, D.V. Molecular Structures of Tetranuclear (Al, Fe) Metal Clusters. Glass Phys. Chem. 2018, 44, 339–345. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Molekularnye struktury tetrayadernykh (Al, Fe) metalloklasterov. Fizika i Khimiya Stekla 2018, 44, 408–415. [Google Scholar]
- Chachkov, D.V.; Mikhailov, O.V. Molekulyarnye struktury poliyadernykh metalloklasterov po dannym raschyota metodom DFT. (Geterotri)tetrayadernyi klaster Al2FeCo. Herald Technol. Univ. 2016, 19, 30–32. [Google Scholar]
- Mikhailov, O.V.; Chachkov, D.V. Quantum Chemical Calculation of Molecular Structures of Al2Fe2 and Al2FeCo Tetranuclear Metalloclusters. Glass Phys. Chem. 2017, 43, 597–604. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Kvantovo-khimicheskii raschyot molekularnykh struktur tetrayadernykh metalloklasterov Al2Fe2 i Al2FeCo. Fizika i Khimiya Stekla 2017, 43, 632–639. [Google Scholar]
- Chachkov, D.V.; Mikhailov, O.V. Molekulyarnye struktury poliyadernykh metalloklasterov po dannym raschyota metodom DFT. (Geterobi)pentayadernyi klaster Al2Ti3. Herald Technol. Univ. 2016, 19, 5–8. [Google Scholar]
- Chachkov, D.V.; Mikhailov, O.V. Molekulyarnye struktury poliyadernykh metalloklasterov po dannym raschyota metodom DFT. (Geterobi)pentayadernyi klaster Al2V3. Herald Technol. Univ. 2016, 19, 12–15. [Google Scholar]
- Mikhailov, O.V.; Chachkov, D.V. Molecular structure models of Al2Ti3 and Al2V3 clusters according to DFT quantum-chemical calculation. Eur. Chem. Bull. 2020, 9, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Mikhailov, O.V.; Chachkov, D.V. Models of Molecular Structures of Al2Cr3 and Al2Mo3 Metal Clusters according to Density Functional Theory Calculations. Russ. J. Inorg. Chem. 2018, 63, 786–799. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Modeli molekularnykh struktur metalloklasterov Al2Cr3 i Al2Mo3 po dannym metoda funktsionala plotnosti. Zh. Neorg. Khim. 2018, 63, 750–763. [Google Scholar]
- Mikhailov, O.V.; Chachkov, D.V. Quantum-chemical calculation of molecular structures of Al2Mn3 and Al2Zn3 clusters by using DFT method. Struct. Chem. 2019, 30, 1289–1299. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Models of Molecular Structure of Heteronuclear Clusters Al2Fe3, Al2Co3, and Al2Ni3 According to the Data of Quantum-Chemical Density Functional Simulation. Russ. J. Gen. Chem. 2016, 86, 1991–1998. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Modeli molekularnykh struktur geteroyadernykh klasterov Al2Fe3, Al2Co3 и Al2Ni3 po dannym kvantovo-khimicheskogo raschyota po metody funktsionala plotnosti. Zh. Obshch. Khim. 2016, 86, 1419–1428. [Google Scholar]
- Mikhailov, O.V.; Chachkov, D.V. DFT calculation of molecular structures of Al2Fe3 and Al2Cu3 heterobinuclear clusters. Struct. Chem. 2018, 29, 1543–1549. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. DFT Quantum Chemical Calculation of the Molecular Structures of the Metal Clusters Al2Cu3 and Al2Ag3. Russ. J. Inorg. Chem. 2019, 64, 79–87. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. DFT Kvantovo-khimicheskii raschyot molekularnykh struktur metalloklasterov Al2Cu3 i Al2Ag3 metodom DFT. Zh. Neorg. Khim. 2019, 64, 63–71. [Google Scholar]
- Mikhailov, O.V.; Chachkov, D.V. Thermodynamics of Al2M3 Metal Clusters (M = 3d-element) in the framework of quantum-chemical modeling by DFT method. Russ. J. Inorg. Chem. 2020, 65, 646–649. [Google Scholar]
- Mikhailov, O.V.; Chachkov, D.V. Termodinamika metalloklasterov Al2M3 (M = 3d-element) v ramkakh kvantovo-khimicheskogo modelirovaniya metodom DFT. Zh. Neorg. Khim. 2020, 65, 598–602. [Google Scholar]
- Chachkov, D.V.; Mikhailov, O.V. Molekulyarnye struktury poliyadernykh metalloklasterov po dannym raschyota metodom DFT. (Geterobi)hexayadernyi klaster Al3Fe3. Herald Technol. Univ. 2016, 19, 89–93. [Google Scholar]
- Chachkov, D.V.; Mikhailov, O.V. Molecular Structure of Hexatomic Heteronuclear (AlFe) Metal Clusters as Determined by the DFT Quantum-Chemical Calculation. Russ. J. Gen. Chem. 2017, 87, 670–678. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. Molekulyarnye struktury shestiatomnykh geteroyadernykh (AlFe) metalloklasterov po dannym po dannym kvantovo-khimicheskogo raschyota metodom DFT. Zh. Obshch. Khim. 2017, 87, 535–543. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure Designation | Spin Multiplicity of the Ground State | Relative Energy, kJ/mol | Ref. |
|---|---|---|---|
| Al3Fe Metal Cluster | |||
| Al3Fe (I) | 2 | 0.0 | [79,80] |
| Al3Fe (VII) | 2 | 175.1 | |
| Al3Fe (II) | 4 | 32.7 | |
| Al3Fe (IV) | 4 | 153.3 | |
| Al3Fe (III) | 6 | 83.4 | |
| Al3Fe (VI) | 6 | 84.8 | |
| Al3Fe (V) | 6 | 193.1 | |
| AlFe3 Metal Cluster | |||
| AlFe3 (II) | 2 | 11.2 | [79,80] |
| AlFe3 (I) | 2 | 104.0 | |
| AlFe3 (VII) | 2 | 122.8 | |
| AlFe3 (VIII) | 2 | 198.7 | |
| AlFe3 (V) | 4 | 0.0 | |
| AlFe3 (III) | 4 | 11.3 | |
| AlFe3 (IX) | 4 | 150.2 | |
| AlFe3 (VI) | 6 | 17.4 | |
| AlFe3 (IV) | 6 | 41.7 | |
| AlFe3 (X) | 6 | 52.6 | |
| Al2Fe2 Metal Cluster | |||
| Al2Fe2 (XII) | 1 | 45.1 | [78,79,80] |
| Al2Fe2 (VII) | 1 | 46.7 | |
| Al2Fe2 (IX) | 1 | 150.3 | |
| Al2Fe2 (X) | 1 | 209.8 | |
| Al2Fe2 (III) | 1 | 352.5 | |
| Al2Fe2 (VI) | 3 | 66.5 | |
| Al2Fe2 (VIII) | 3 | 68.4 | |
| Al2Fe2 (IV) | 3 | 137.9 | |
| Al2Fe2 (XI) | 3 | 143.6 | |
| Al2Fe2 (II) | 3 | 254.5 | |
| Al2Fe2 (V) | 5 | 0.0 | |
| Al2Fe2 (I) | 5 | 152.7 | |
| Al3Fe Metal Cluster | Al2Fe2 Metal Cluster | AlFe3 Metal Cluster | |||
|---|---|---|---|---|---|
| Metal-Metal Bond Lengths, pm | Metal-Metal Bond Lengths, pm | Metal-Metal Bond Lengths, pm | |||
| Al1Al2 | 263.4 | Al1Al2 | 260.8 | Al1Fe1 | 249.1 |
| Al1Al3 | 263.3 | Al1Fe1 | 249.5 | Al1Fe2 | 249.1 |
| Al2Al3 | 274.3 | Al1Fe2 | 249.4 | Al1Fe3 | 255.5 |
| Al1Fe1 | 245.7 | Al2Fe1 | 249.5 | Fe1Fe2 | 208.6 |
| Al2Fe1 | 235.0 | Al2Fe2 | 249.5 | Fe1Fe3 | 248.7 |
| Al3Fe1 | 235.1 | Fe1Fe2 | 199.2 | Fe2Fe3 | 248.7 |
| Bond Angles, deg | Bond Angles, deg | Bond Angles, deg | |||
| Al1Fe1Al2 | 66.4 | Fe1Al1Fe2 | 47.1 | Fe1Al1Fe2 | 49.5 |
| Fe1Al1Al2 | 54.9 | Fe1Al2Fe2 | 47.1 | Al1Fe1Fe2 | 65.3 |
| Al1Al2Fe1 | 58.7 | Fe1Al1Al2 | 58.5 | Fe1Fe2Al1 | 65.2 |
| Al1Fe1Al3 | 66.4 | Fe1Al2Al1 | 58.5 | Fe1Al1Fe3 | 59.0 |
| Al2Fe1Al3 | 71.4 | Fe2Al1Al2 | 58.5 | Fe2Al1Fe3 | 59.0 |
| Al1Al2Al3 | 58.6 | Fe2Al2Al1 | 58.5 | Fe1Fe2Fe3 | 65.2 |
| Al2Al3Al1 | 58.6 | Al1Fe1Al2 | 63.0 | Fe2Fe3Fe1 | 49.6 |
| Al3Al1Al2 | 62.8 | Al1Fe2Al2 | 63.1 | Fe3Fe1Fe2 | 65.2 |
| M | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn |
|---|---|---|---|---|---|---|---|---|---|
| N | 14 | 11 | 20 | 25 | 8 | 9 | 7 | 8 | 14 |
| Ref. | [81,83] | [82,83] | [84,85] | [86] | [87,88,89] | [87,88] | [87,88] | [89,90,91] | [86] |
| Structure Designation | Spin Multiplicity of the Ground State | Relative Energy, kJ/mol | Ref. |
|---|---|---|---|
| Al2Ti3 Metal Cluster | |||
| Al2Ti3 (XIII) | 1 | 21.5 | [81,83] |
| Al2Ti3 (II) | 1 | 24.1 | |
| Al2Ti3 (VII) | 1 | 44.8 | |
| Al2Ti3 (XIV) | 1 | 51.1 | |
| Al2Ti3 (VIII) | 1 | 77.5 | |
| Al2Ti3 (III) | 1 | 93.0 | |
| Al2Ti3 (XII) | 3 | 12.6 | |
| Al2Ti3 (X) | 3 | 37.0 | |
| Al2Ti3 (VI) | 3 | 37.2 | |
| Al2Ti3 (I) | 3 | 80.9 | |
| Al2Ti3 (XI) | 5 | 0.0 | |
| Al2Ti3 (V) | 5 | 19.7 | |
| Al2Ti3 (IV) | 5 | 60.7 | |
| Al2Ti3 (IX) | 5 | 73.0 | |
| Al2V3 Metal Cluster | |||
| Al2V3 (I) | 2 | 25.9 | [82,83] |
| Al2V3 (IV) | 2 | 26.7 | |
| Al2V3 (VII) | 2 | 30.2 | |
| Al2V3 (V) | 4 | 0.0 | |
| Al2V3 (II) | 4 | 2.4 | |
| Al2V3 (X) | 4 | 59.6 | |
| Al2V3 (VIII) | 4 | 71.3 | |
| Al2V3 (III) | 6 | 18.8 | |
| Al2V3 (VI) | 6 | 26.8 | |
| Al2V3 (XI) | 6 | 74.6 | |
| Al2V3 (IX) | 6 | 141.0 | |
| Al2Cr3 Metal Cluster | |||
| Al2Cr3 (XVIII) | 1 | 172.6 | [84,85] |
| Al2Cr3 (XVI) | 1 | 186.5 | |
| Al2Cr3 (V) | 1 | 197.2 | |
| Al2Cr3 (XIII) | 1 | 206.8 | |
| Al2Cr3 (X) | 1 | 219.0 | |
| Al2Cr3 (XIX) | 1 | 266.6 | |
| Al2Cr3 (VI) | 1 | 287.0 | |
| Al2Cr3 (I) | 1 | 396.2 | |
| Al2Cr3 (VII) | 3 | 79.4 | |
| Al2Cr3 (II) | 3 | 92.9 | |
| Al2Cr3 (XIV) | 3 | 109.3 | |
| Al2Cr3 (XI) | 3 | 109.6 | |
| Al2Cr3 (III) | 5 | 0.0 | |
| Al2Cr3 (XVII) | 5 | 13.1 | |
| Al2Cr3 (VIII) | 5 | 19.5 | |
| Al2Cr3 (XII) | 5 | 39.6 | |
| Al2Cr3 (XX) | 5 | 73.4 | |
| Al2Cr3 (XV) | 5 | 86.8 | |
| Al2Cr3 (IV) | 7 | 22.9 | |
| Al2Cr3 (IX) | 7 | 39.6 | |
| Al2Mn3 Metal Cluster | |||
| Al2Mn3 (XIII) | 2 | 29.3 | [86] |
| Al2Mn3 (XVII) | 2 | 34.9 | |
| Al2Mn3 (V) | 2 | 47.5 | |
| Al2Mn3 (XXII) | 2 | 50.5 | |
| Al2Mn3 (XXV) | 2 | 80.8 | |
| Al2Mn3 (VIII) | 2 | 124.0 | |
| Al2Mn3 (XIX) | 2 | 129.4 | |
| Al2Mn3 (XXI) | 4 | 2.2 | |
| Al2Mn3 (XII) | 4 | 5.9 | |
| Al2Mn3 (IV) | 4 | 11.2 | |
| Al2Mn3 (XVI) | 4 | 27.0 | |
| Al2Mn3 (VII) | 4 | 29.6 | |
| Al2Mn3 (XXIV) | 4 | 76.1 | |
| Al2Mn3 (II) | 4 | 82.2 | |
| Al2Mn3 (X) | 4 | 141.7 | |
| Al2Mn3 (VI) | 6 | 0.0 | |
| Al2Mn3 (XX) | 6 | 1.4 | |
| Al2Mn3 (III) | 6 | 19.1 | |
| Al2Mn3 (XV) | 6 | 28.1 | |
| Al2Mn3 (XI) | 6 | 36.6 | |
| Al2Mn3 (XIV) | 6 | 42.3 | |
| Al2Mn3 (XXIII) | 6 | 62.4 | |
| Al2Mn3 (IX) | 6 | 74.6 | |
| Al2Mn3 (I) | 6 | 77.2 | |
| Al2Mn3 (XVIII) | 6 | 149.2 | |
| Al2Fe3 Metal Cluster | |||
| Al2Fe3(I) | 1 | 273.7 | [87,88,89] |
| Al2Fe3(IV) | 1 | 300.6 | |
| Al2Fe3(II) | 3 | 0.0 | |
| Al2Fe3(V) | 3 | 24.1 | |
| Al2Fe3(VII) | 3 | 27.1 | |
| Al2Fe3(III) | 5 | 12.8 | |
| Al2Fe3(VIII) | 5 | 24.7 | |
| Al2Fe3(VI) | 5 | 31.1 | |
| Al2Co3 Metal Cluster | |||
| Al2Co3(I) | 2 | 56.0 | [87,88] |
| Al2Co3(VII) | 2 | 83.6 | |
| Al2Co3(IV) | 2 | 92.0 | |
| Al2Co3(II) | 4 | 86.1 | |
| Al2Co3(VIII) | 4 | 86.8 | |
| Al2Co3(V) | 4 | 96.3 | |
| Al2Co3(III) | 6 | 0.0 | |
| Al2Co3(IX) | 6 | 28.9 | |
| Al2Co3(VI) | 6 | 73.3 | |
| Al2Ni3 Metal Cluster | |||
| Al2Ni3 (I) | 1 | 48.6 | [87,88] |
| Al2Ni3 (IV) | 1 | 70.8 | |
| Al2Ni3 (II) | 3 | 0.0 | |
| Al2Ni3 (V) | 3 | 73.1 | |
| Al2Ni3 (III) | 5 | 102.6 | |
| Al2Ni3 (VI) | 5 | 113.3 | |
| Al2Ni3 (VII) | 5 | 148.4 | |
| Al2Cu3 Metal Cluster | |||
| Al2Cu3 (I) | 2 | 0.0 | [89,90,91] |
| Al2Cu3 (III) | 2 | 20.6 | |
| Al2Cu3 (V) | 2 | 27.0 | |
| Al2Cu3 (VII) | 2 | 40.1 | |
| Al2Cu3 (VIII) | 2 | 67.6 | |
| Al2Cu3 (IV) | 4 | 136.9 | |
| Al2Cu3 (II) | 4 | 144.1 | |
| Al2Cu3 (VI) | 4 | 144.3 | |
| Al2Zn3 Metal Cluster | |||
| Al2Zn3 (III) | 1 | 14.0 | [86] |
| Al2Zn3 (II) | 1 | 17.7 | |
| Al2Zn3 (XII) | 1 | 18.9 | |
| Al2Zn3 (VI) | 1 | 22.1 | |
| Al2Zn3 (XIII) | 1 | 23.8 | |
| Al2Zn3 (XI) | 1 | 25.6 | |
| Al2Zn3 (V) | 1 | 29.6 | |
| Al2Zn3 (IX) | 1 | 30.5 | |
| Al2Zn3 (VIII) | 1 | 79.2 | |
| Al2Zn3 (I) | 3 | 0.0 | |
| Al2Zn3 (XIV) | 3 | 3.2 | |
| Al2Zn3 (IV) | 3 | 11.0 | |
| Al2Zn3 (VII) | 3 | 21.4 | |
| Al2Zn3 (X) | 3 | 29.6 | |
| M | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn | |
|---|---|---|---|---|---|---|---|---|---|---|
| Parameter | ||||||||||
| Metal–Metal Bond Lengths, pm | ||||||||||
| Al1Al2 | 422.7 | 270.2 | 262.4 | 276.1 | 273.9 | 271.5 | 270.7 | 271.1 | 277.3 | |
| Al1M1 | 254.4 | 263.7 | 262.6 | 260.4 | 244.8 | 233.1 | 229.0 | 244.1 | 267.7 | |
| Al1M2 | 258.2 | 265.7 | 278.1 | 257.6 | 240.9 | 252.8 | 236.1 | 254.2 | 267.8 | |
| Al1M3 | 258.2 | 252.2 | 262.6 | 260.4 | 252.3 | 233.1 | 229.0 | 244.1 | 273.7 | |
| Al2M1 | 254.4 | 261.0 | 262.6 | 260.4 | 244.8 | 233.1 | 229.0 | 244.1 | 267.7 | |
| Al2M2 | 258.2 | 378.8 | 278.1 | 257.7 | 240.9 | 252.8 | 236.1 | 254.2 | 247.9 | |
| Al2M3 | 258.2 | 254.4 | 262.6 | 260.4 | 252.3 | 233.1 | 229.0 | 244.1 | 273.6 | |
| M1M3 | 258.7 | 265.4 | 415.9 | 434.8 | 250.7 | 341.6 | 357.3 | 384.1 | 446.0 | |
| M2M3 | 239.0 | 253.3 | 258.1 | 281.6 | 217.6 | 215.3 | 231.1 | 243.1 | 446.1 | |
| M1M2 | 258.8 | 171.7 | 258.1 | 281.6 | 374.9 | 215.3 | 231.1 | 243.1 | 247.9 | |
| Bond Angles, deg | ||||||||||
| M1Al1M2 | 60.6 | 37.8 | 56.9 | 65.9 | 101.0 | 52.4 | 59.6 | 58.3 | 55.2 | |
| M1Al2M2 | 60.6 | 22.9 | 56.9 | 65.8 | 101.0 | 52.4 | 59.6 | 58.3 | 55.2 | |
| M1Al1Al2 | 33.8 | 58.5 | 60.0 | 58.0 | 56.0 | 54.4 | 53.8 | 56.3 | 58.8 | |
| M1Al2Al1 | 33.8 | 59.5 | 60.0 | 58.0 | 56.0 | 54.4 | 53.8 | 56.3 | 58.8 | |
| M2Al1Al2 | 35.6 | 90.0 | 61.9 | 57.6 | 55.4 | 57.5 | 55.0 | 57.8 | 58.8 | |
| M2Al2Al1 | 35.0 | 44.5 | 61.9 | 57.6 | 55.4 | 57.5 | 55.0 | 57.8 | 58.8 | |
| Al1M1Al2 | 112.3 | 62.0 | 59.9 | 64.0 | 68.0 | 71.2 | 72.5 | 67.4 | 62.4 | |
| Al1M2Al2 | 109.9 | 45.5 | 56.3 | 64.8 | 69.3 | 64.9 | 69.9 | 64.4 | 62.4 | |
| Al1M3Al2 | 109.9 | 64.5 | 59.9 | 64.0 | 65.7 | 68.5 | 72.5 | 67.4 | 60.9 | |
| M1Al1M3 | 60.6 | 61.9 | 104.7 | 113.2 | 60.5 | 94.2 | 102.6 | 103.7 | 110.9 | |
| M1Al2M3 | 60.6 | 62.0 | 104.7 | 113.2 | 60.5 | 94.2 | 102.6 | 103.7 | 111.0 | |
| M1M3M2 | 62.5 | 38.6 | 36.3 | 65.9 | 106.2 | 37.5 | 39.4 | 37.8 | 32.3 | |
| M2Al1M3 | 55.1 | 58.5 | 56.9 | 65.9 | 52.3 | 52.4 | 59.6 | 58.3 | 110.9 | |
| M2Al2M3 | 55.1 | 41.6 | 56.9 | 101.1 | 52.3 | 52.4 | 59.6 | 58.3 | 111.0 | |
| M1M2M3 | 62.5 | 74.5 | 107.3 | 39.5 | 40.0 | 105.0 | 101.3 | 104.4 | 73.9 | |
| Metal Cluster | Standard Thermodynamic Parameters of Formation | ||
|---|---|---|---|
| ΔfH0(298 K) kJ/mol | ΔfS0(298 K) J/mol∙ K | ΔfG0(298 K) kJ/mol | |
| Al2Ti3 | 967.4 | 429.9 | 883.5 |
| Al2V3 | 526.5 | 438.5 | 433.8 |
| Al2Cr3 | 1151.1 | 417.8 | 1067.5 |
| Al2Mn3 | 516.8 | 423.1 | 436.2 |
| Al2Fe3 | 823.4 | 430.0 | 736.5 |
| Al2Co3 | 817.6 | 427.6 | 733.9 |
| Al2Ni3 | 760.9 | 430.0 | 676.3 |
| Al2Cu3 | 812.2 | 406.9 | 737.7 |
| Al2Zn3 | 700.1 | 445.1 | 621.7 |
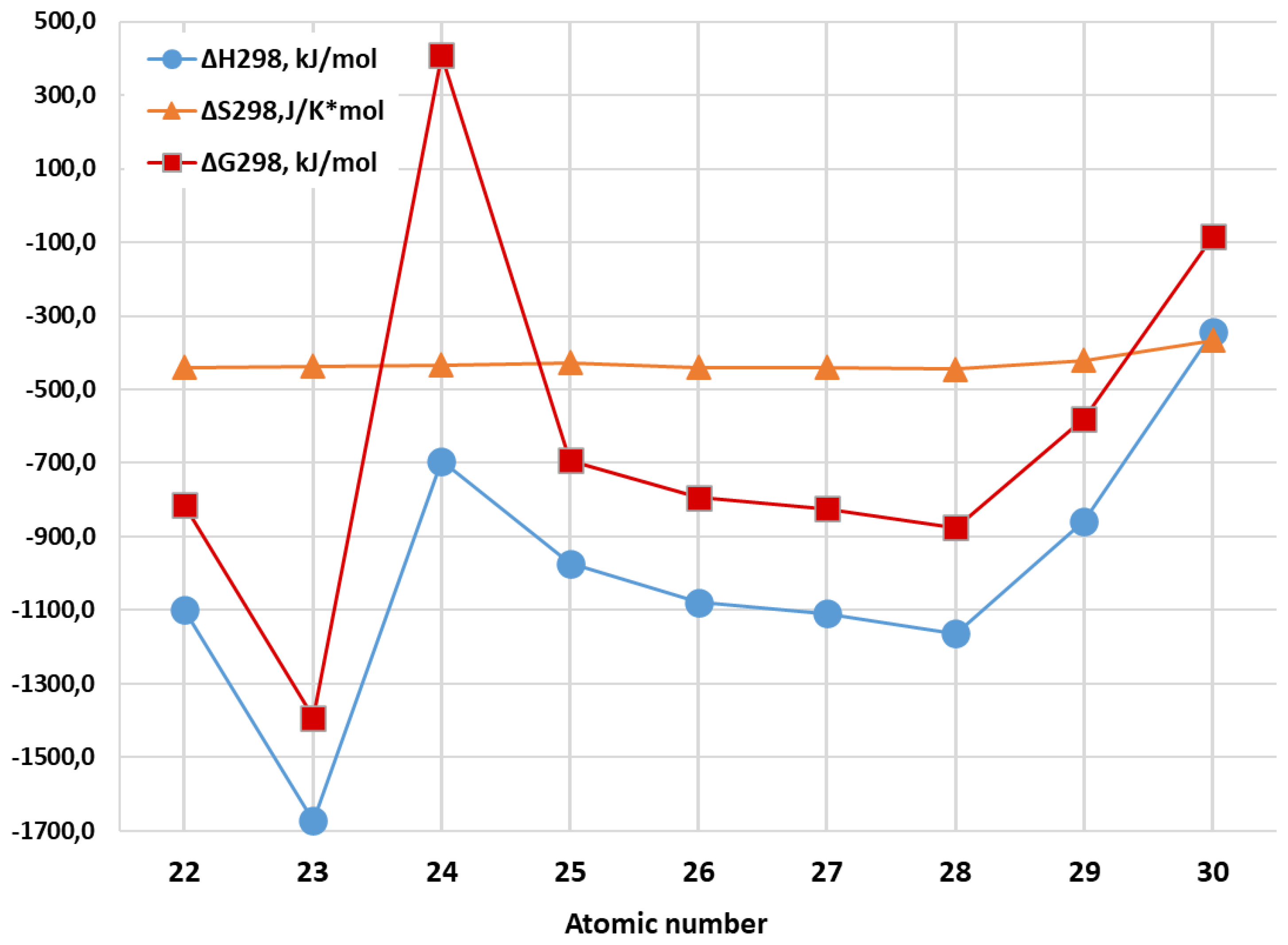
| Metal Cluster | Standard Thermodynamic Parameters of Reactions 2Al(gas) + 3M(gas)→Al2M3 (gas) | ||
| ΔH298, kJ/mol | ΔS298, J/mol∙ K | ΔG298, kJ/mol | |
| Al2Ti3 | –1098.5 | –439.5 | –813.4 |
| Al2V3 | –1672.0 | –436.9 | –1392.1 |
| Al2Cr3 | –694.0 | –433.6 | 407.7 |
| Al2Mn3 | –973.4 | –426.5 | –692.2 |
| Al2Fe3 | –1078.1 | –440.0 | –793.0 |
| Al2Co3 | –1109.1 | –439.4 | –824.1 |
| Al2Ni3 | –1162.8 | –445.1 | –876.0 |
| Al2Cu3 | –858.3 | –420.8 | –578.8 |
| Al2Zn3 | –344.6 | –366.4 | –84.3 |
| Metal Cluster | Atomic Number of M | Ttd, K |
|---|---|---|
| Al2Ti3 | 22 | 2502.2 |
| Al2V3 | 23 | 3826.0 |
| Al2Cr3 | 24 | 1599.0 |
| Al2Mn3 | 25 | 2278.6 |
| Al2Fe3 | 26 | 2450.2 |
| Al2Co3 | 27 | 2526.4 |
| Al2Ni3 | 28 | 2613.0 |
| Al2Cu3 | 29 | 2038.7 |
| Al2Zn3 | 30 | 941.5 |
| Structure Designation | Spin Multiplicity of the Ground State | Relative Energy, kJ/mol | Ref. |
|---|---|---|---|
| Al3Fe3 Metal Cluster | |||
| Al3Fe3 (IV) | 2 | 58.8 | [94,95,96] |
| Al3Fe3 (X) | 2 | 95.2 | |
| Al3Fe3 (XIX) | 2 | 108.3 | |
| Al3Fe3 (VII) | 2 | 134.5 | |
| Al3Fe3 (XIII) | 2 | 137.2 | |
| Al3Fe3 (XVI) | 2 | 153.7 | |
| Al3Fe3 (I) | 2 | 158.3 | |
| Al3Fe3 (V) | 4 | 56.8 | |
| Al3Fe3 (XIV) | 4 | 56.8 | |
| Al3Fe3 (VIII) | 4 | 108.9 | |
| Al3Fe3 (XI) | 4 | 110.1 | |
| Al3Fe3 (II) | 4 | 130.8 | |
| Al3Fe3 (XV) | 6 | 0.0 | |
| Al3Fe3 (XII) | 6 | 40.8 | |
| Al3Fe3 (IX) | 6 | 71.8 | |
| Al3Fe3 (VI) | 6 | 77.3 | |
| Al3Fe3 (III) | 6 | 78.1 | |
| Al3Fe3 (XX) | 6 | 105.9 | |
| Al3Fe3 (XVII) | 6 | 111.4 | |
| Al3Fe3 (XVIII) | 6 | 155.4 | |
| Al2Fe4 Metal Cluster | |||
| Al2Fe4 (V) | 3 | 13.0 | [95,96] |
| Al2Fe4 (VIII) | 3 | 27.5 | |
| Al2Fe4 (III) | 3 | 80.8 | |
| Al2Fe4 (IX) | 3 | 102.8 | |
| Al2Fe4 (VI) | 3 | 115.7 | |
| Al2Fe4 (I) | 3 | 165.5 | |
| Al2Fe4 (II) | 5 | 0.0 | |
| Al2Fe4 (VII) | 5 | 25.5 | |
| Al2Fe4 (IV) | 5 | 79.2 | |
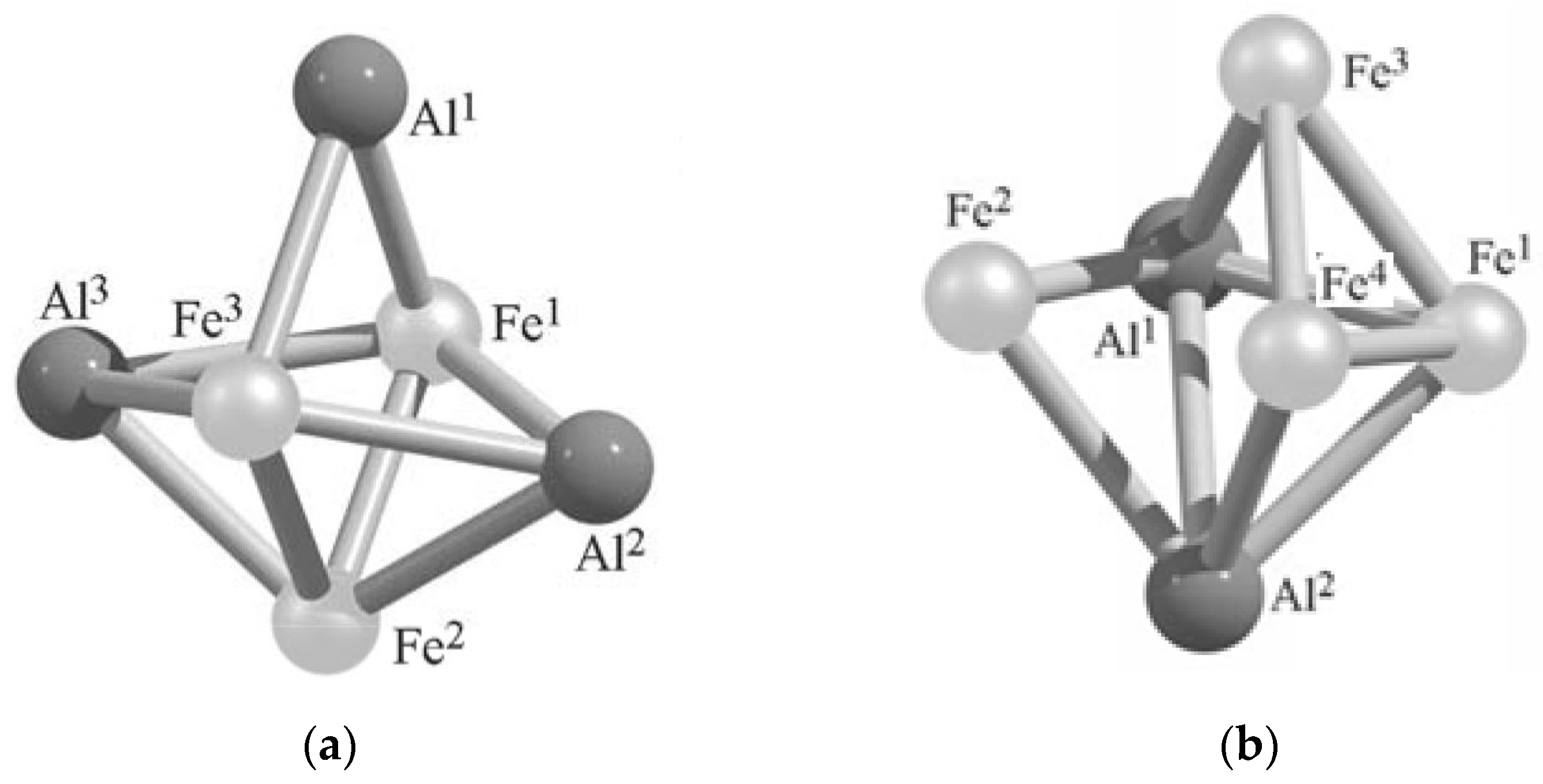
| Al3Fe3 Metal Cluster | Al2Fe4 Metal Cluster | ||
|---|---|---|---|
| Metal–Metal Bond Lengths, pm | Metal–Metal Bond Lengths, pm | ||
| Al1Al2 | 306.2 | Al1Fe1 | 242.7 |
| Al1Al3 | 307.1 | Al1Fe2 | 249.8 |
| Al2Al3 | 398.9 | Al1Fe3 | 243.8 |
| Al1Fe1 | 236.4 | Al1Fe4 | 350.4 |
| Al1Fe2 | 370.1 | Al1Al2 | 274.7 |
| Al1Fe3 | 236.3 | Al2Fe1 | 242.7 |
| Al2Fe1 | 244.4 | Al2Fe2 | 249.7 |
| Al2Fe2 | 242.1 | Al2Fe3 | 350.4 |
| Al2Fe3 | 244.2 | Al2Fe4 | 243.8 |
| Al3Fe1 | 244.3 | Fe2Fe3 | 244.1 |
| Al3Fe2 | 242.1 | Fe1Fe4 | 225.9 |
| Al3Fe3 | 244.2 | Fe1Fe3 | 225.9 |
| Fe1Fe2 | 222.5 | Fe1Fe2 | 329.1 |
| Fe1Fe3 | 270.9 | Fe2Fe4 | 244.1 |
| Fe2Fe3 | 222.4 | Fe3Fe4 | 230.7 |
| Bond Angles, deg | Bond Angles, deg | ||
| Fe2Al1Fe3 | 35.0 | Fe2Al1Fe3 | 59.3 |
| Fe1Al1Fe2 | 35.0 | Fe1Al1Fe2 | 83.9 |
| Fe1Al2Al3 | 35.3 | Fe1Al2Fe4 | 55.4 |
| Fe2Al2Al3 | 34.5 | Fe2Al2Fe4 | 59.3 |
| Al1Al2Fe2 | 84.1 | Al1Al2Fe2 | 56.6 |
| Fe2Al1Al2 | 40.6 | Fe2Al1Al2 | 56.6 |
| Al1Al2Al3 | 49.5 | Al1Al2Fe4 | 84.8 |
| Al2Al3Fe1 | 35.3 | Al2Fe4Fe1 | 62.1 |
| Al3Fe1Al2 | 109.4 | Fe4Fe1Al2 | 62.6 |
| Fe3Al1Al2 | 51.6 | Fe3Al1Al2 | 84.8 |
| Fe1Al1Fe3 | 69.9 | Fe1Al1Fe3 | 55.4 |
| Fe1Al1Al2 | 51.6 | Fe1Al1Al2 | 55.5 |
| Al1Fe1Al2 | 79.1 | Al1Fe1Al2 | 68.9 |
| Al3Fe2Fe3 | 63.3 | Fe4Fe2Fe3 | 56.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikhailov, O.V.; Chachkov, D.V. Quantum-Chemical Design of Molecular Structures of Tetra-, Penta- and Hexanuclear Metal Clusters Containing Aluminum and 3d-Element Atoms. Materials 2020, 13, 1852. https://doi.org/10.3390/ma13081852
Mikhailov OV, Chachkov DV. Quantum-Chemical Design of Molecular Structures of Tetra-, Penta- and Hexanuclear Metal Clusters Containing Aluminum and 3d-Element Atoms. Materials. 2020; 13(8):1852. https://doi.org/10.3390/ma13081852
Chicago/Turabian StyleMikhailov, Oleg V., and Denis V. Chachkov. 2020. "Quantum-Chemical Design of Molecular Structures of Tetra-, Penta- and Hexanuclear Metal Clusters Containing Aluminum and 3d-Element Atoms" Materials 13, no. 8: 1852. https://doi.org/10.3390/ma13081852