Characterization of PLA/PBSeT Blends Prepared with Various Hexamethylene Diisocyanate Contents

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of PBSeT and Blending
2.3. Specimen Preparation
2.4. Fourier-Transform Infrared Analysis
2.5. Mechanical-Property Analysis
2.6. Gel-Permeation-Chromatography Analysis
2.7. Hydrolytic Degradation Measurements
2.8. Thermal-Property Analysis
3. Results and Discussion
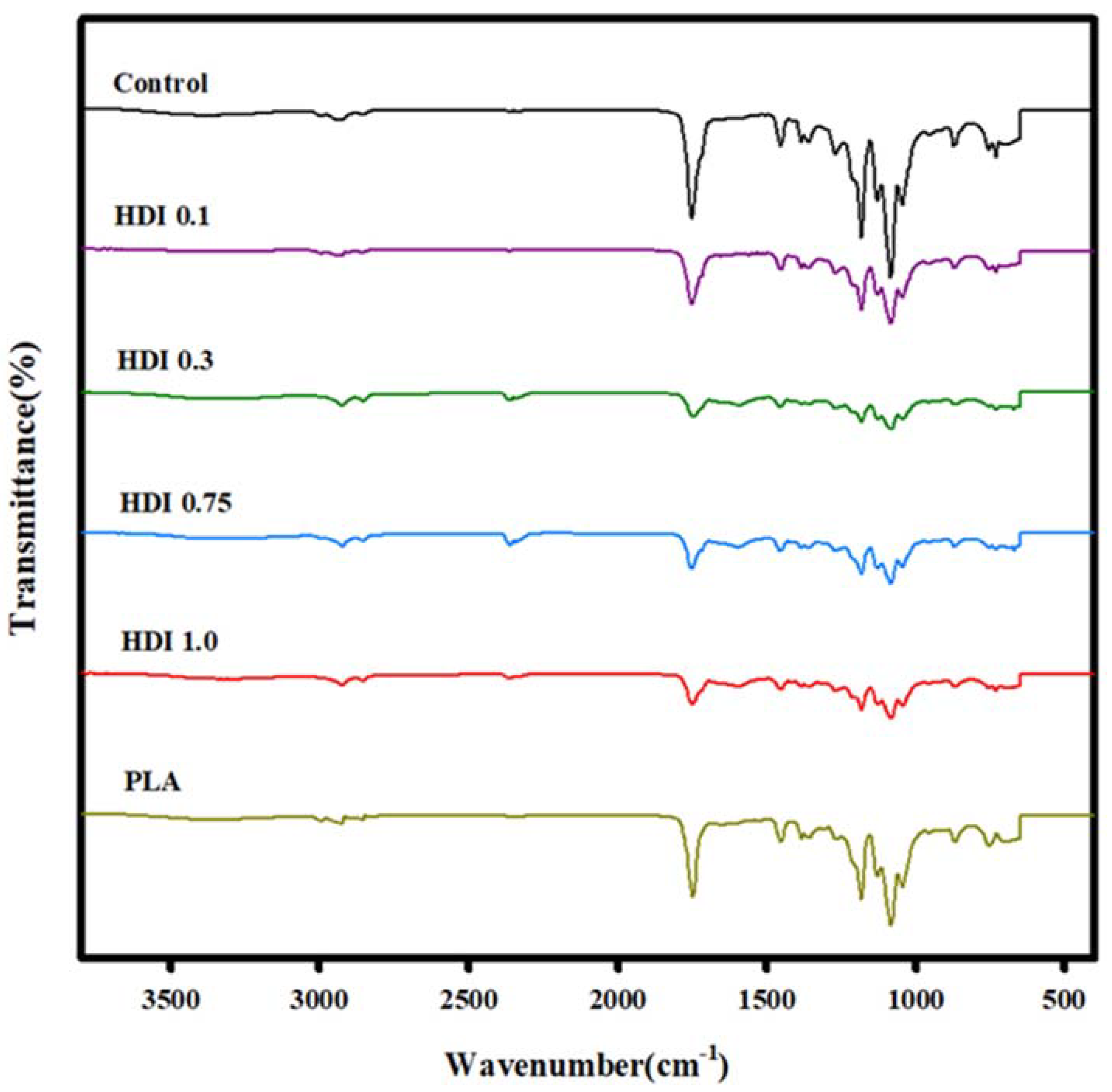
3.1. FTIR Analysis
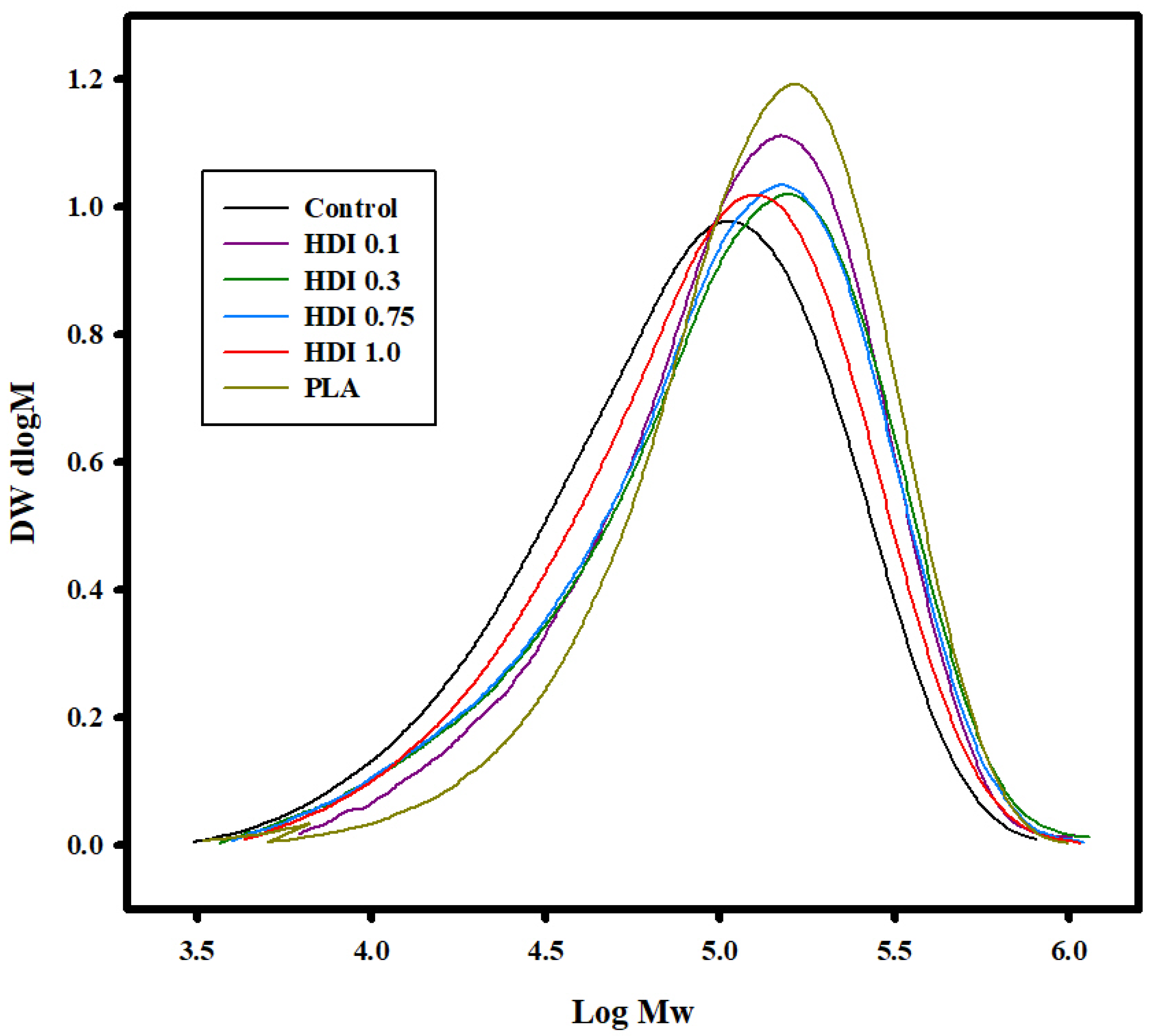
3.2. GPC Analysis
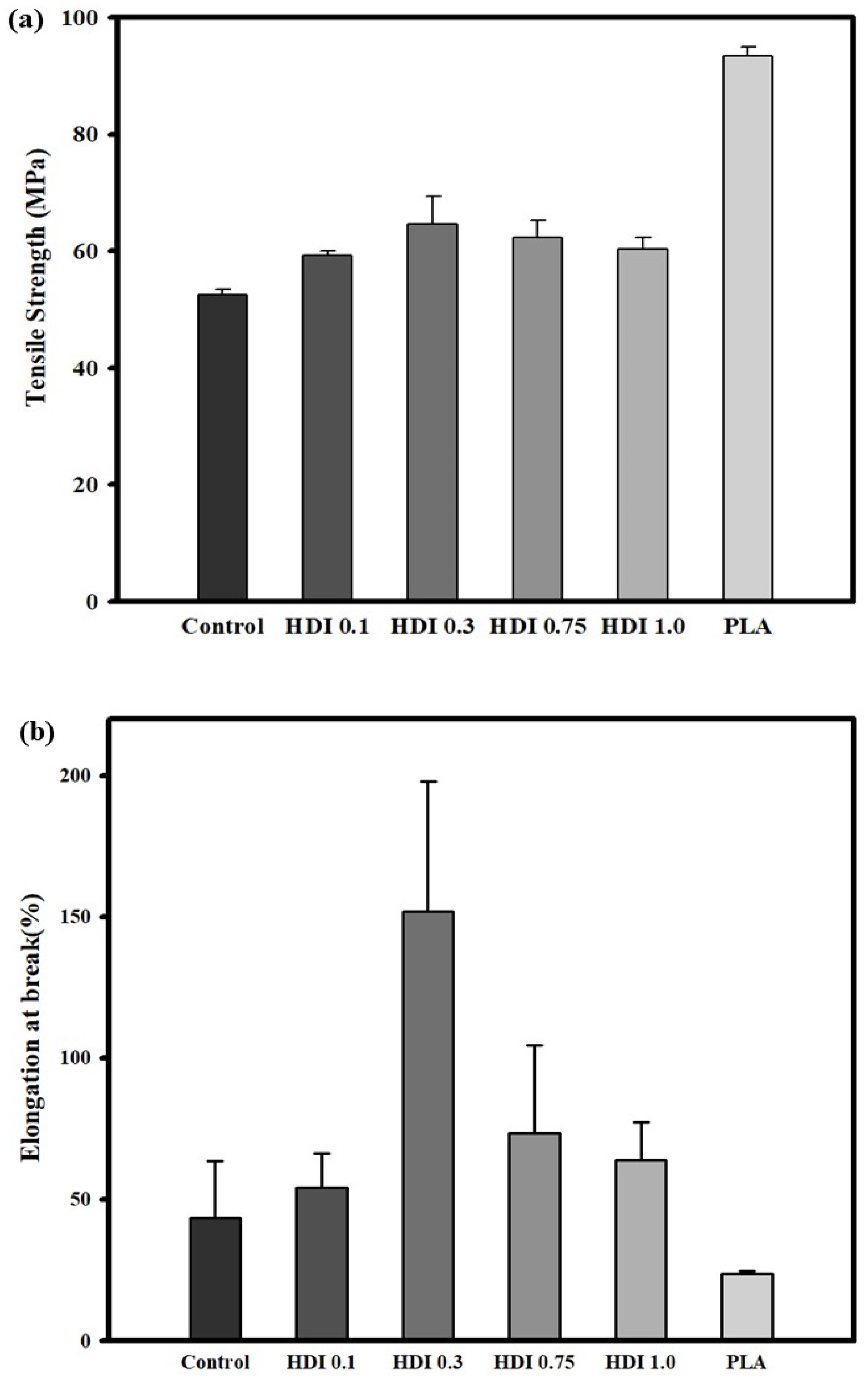
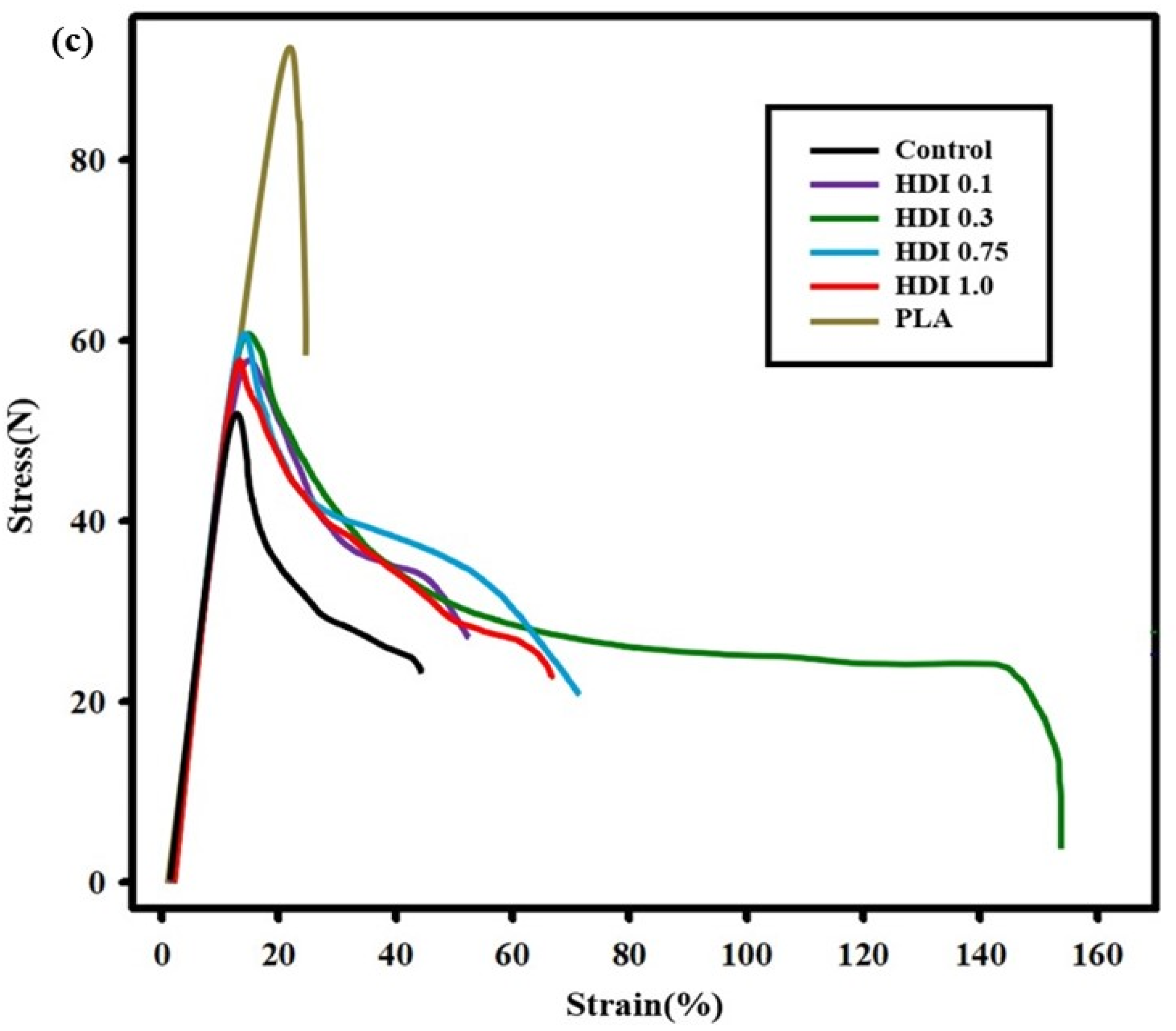
3.3. Tensile Strength and Elongation Properties
3.4. Three-Point Flexural Strengths
3.5. Thermal Properties
3.6. Hydrolytic Degradation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yu, L.; Dean, K.; Li, L. Polymer blends and composites from renewable resources. Prog. Polym. Sci. 2006, 31, 576–602. [Google Scholar] [CrossRef]
- Dilkes-Hoffman, L.S.; Pratt, S.; Lant, P.A.; Laycock, B. The role of biodegradable plastic in solving plastic solid waste accumulation. In Plastics to Energy: Fuel, Chemicals, and Sustainability Implications, 1st ed.; Al-Salem, S.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 469–505. ISBN 978-0-12-813140-4. [Google Scholar] [CrossRef]
- Van den Oever, M.; Molenveld, K. Replacing fossil based plastic performance products by bio-based plastic products—technical feasibility. New Biotechnol. 2017, 37, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Varga, N.; Turcsânyi, Á.; Hornok, V.; Csapó, E. Vitamin e-loaded pla- and plga-based core-shell nanoparticles: Synthesis, structure optimization and controlled drug release. Pharmaceutics 2019, 11, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, W.; Mehboob, A.; Han, M.G.; Chang, S.H. Effect of fluoride coating on degradation behaviour of unidirectional Mg/PLA biodegradable composite for load-bearing bone implant application. Compos. Part A Appl. Sci. Manuf. 2019, 124, 105464. [Google Scholar] [CrossRef]
- Gregor, A.; Filová, E.; Novák, M.; Kronek, J.; Chlup, H.; Buzgo, M.; Blahnová, V.; Lukášová, V.; Bartoš, M.; Nečas, A.; et al. Designing of PLA scaffolds for bone tissue replacement fabricated by ordinary commercial 3D printer. J. Biol. Eng. 2017, 11, 31. [Google Scholar] [CrossRef]
- Casalini, T.; Rossi, F.; Castrovinci, A.; Perale, G. A perspective on polylactic acid-based polymers use for nanoparticles synthesis and applications. Front. Bioeng. Biotechnol. 2019, 7, 259. [Google Scholar] [CrossRef]
- Friné, V.C.; Hector, A.P.; Manuel, N.D.S.; Estrella, N.D.; Antonio, G.J. Development and characterization of a biodegradable PLA food packaging hold monoterpene-cyclodextrin complexes against Alternaria alternata. Polymers 2019, 11, 1720. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Tang, H.; Wang, Y.; Liu, Y.; Wang, J.; Shi, W.; Li, L. Development of PLA-PBSA based biodegradable active film and its application to salmon slices. Food Packag. Shelf Life 2019, 22, 100393. [Google Scholar] [CrossRef]
- Villegas, C.; Arrieta, M.P.; Rojas, A.; Torres, A.; Faba, S.; Toledo, M.J.; Gutierrez, M.A.; Zavalla, E.; Romero, J.; Galotto, M.J.; et al. PLA/organoclay bionanocomposites impregnated with thymol and cinnamaldehyde by supercritical impregnation for active and sustainable food packaging. Compos. Part B Eng. 2019, 176, 107336. [Google Scholar] [CrossRef]
- Tripathi, P.; Yadav, K. Hybrid bamboo and glass fiber polymer composite-a review. Int. J. Innov. Res. Adv. Eng. 2017, 4, 37–41. [Google Scholar]
- Thapa, S. 3D Printing of Automobile Power Transmission System Using Tough PLA. Master of Science in Materials Processing Technology, Arcada University of Applied Sciences, Helsinki, Finland, 16 April 2020. [Google Scholar]
- Caminero, M.Á.; Chacón, J.M.; García-Plaza, E.; Núñez, P.J.; Reverte, J.M.; Becar, J.P. Additive manufacturing of PLA-based composites using fused filament fabrication: Effect of graphene nanoplatelet reinforcement on mechanical properties, dimensional accuracy and texture. Polymers 2019, 11, 799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniac, I.; Popescu, D.; Zapciu, A.; Antoniac, A.; Miculescu, F.; Moldovan, H. Magnesium filled polylactic acid (PLA) material for filament based 3D printing. Materials 2019, 12, 719. [Google Scholar] [CrossRef] [Green Version]
- Burzic, I.; Pretschuh, C.; Kaineder, D.; Eder, G.; Smilek, J.; Másilko, J.; Kateryna, W. Impact modification of PLA using biobased biodegradable PHA biopolymers. Eur. Polym. J. 2019, 114, 32–38. [Google Scholar] [CrossRef]
- Siakeng, R.; Jawaid, M.; Ariffin, H.; Sapuan, S.M.; Asim, M.; Saba, N. Natural fiber reinforced polylactic acid composites: A review. Polym. Compos. 2019, 40, 446–463. [Google Scholar] [CrossRef]
- Koh, J.J.; Zhang, X.; He, C. Fully biodegradable poly(lactic acid)/starch blends: A review of toughening strategies. Int. J. Biol. Macromol. 2018, 109, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Nofar, M.; Salehiyan, R.; Ciftci, U.; Jalali, A.; Durmuş, A. Ductility improvements of PLA-based binary and ternary blends with controlled morphology using PBAT, PBSA, and nanoclay. Compos. Part B Eng. 2020, 182, 107661. [Google Scholar] [CrossRef]
- Al-Itry, R.; Lamnawar, K.; Maazouz, A. Rheological, morphological, and interfacial properties of compatibilized PLA/PBAT blends. Rheol. Acta 2014, 53, 501–517. [Google Scholar] [CrossRef]
- Fortelny, I.; Ujcic, A.; Fambri, L.; Slouf, M. Phase structure, compatibility, and toughness of PLA/PCL blends: A review. Front. Mater. 2019, 6, 206. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.S. Influence of chain extender on the rheological and thermal properties of polylactic acid (PLA). Bachelor of Science Degree, University of Groningen, Groningen, The Netherlands, 2019. [Google Scholar]
- Farias da Silva, J.M.; Soares, B.G. Epoxidized cardanol-based prepolymer as promising biobased compatibilizing agent for PLA/PBAT blends. Polym. Test 2020, 93, 106889. [Google Scholar] [CrossRef]
- Su, S.; Kopitzky, R.; Tolga, S.; Kabasci, S. Polylactide (PLA) and its blends with poly(butylene succinate) (PBS): A brief review. Polymers 2019, 11, 1193. [Google Scholar] [CrossRef] [Green Version]
- de Moura, N.K.; Siqueira, I.A.W.B.; de Barros Machado, J.P.; Kido, H.W.; Avanzi, I.R.; Muniz Rennó, A.C.; de Sousa Trichês, E.; Passador, F.R. Production and characterization of porous polymeric membranes of PLA/PCL blends with the addition of hydroxyapatite. J. Compos. Sci. 2019, 3, 45. [Google Scholar] [CrossRef] [Green Version]
- Snowdon, M.R.; Mohanty, A.K.; Misra, M. Miscibility and performance evaluation of biocomposites made from polypropylene/poly(lactic acid)/poly(hydroxybutyrate-cohydroxyvalerate) with a sustainable biocarbon filler. ACS Omega 2017, 2, 6446–6454. [Google Scholar] [CrossRef] [PubMed]
- Peltola, H.; Immonen, K.; Johansson, L.S.; Virkajärvi, J.; Sandquist, D. Influence of pulp bleaching and compatibilizer selection on performance of pulp fiber reinforced PLA biocomposites. J. Appl. Polym. Sci. 2019, 136, 47955. [Google Scholar] [CrossRef]
- Qian, S.; Sheng, K. PLA toughened by bamboo cellulose nanowhiskers: Role of silane compatibilization on the PLA bionanocomposite properties. Compos. Sci. Technol. 2017, 148, 59–69. [Google Scholar] [CrossRef]
- Kilic, N.T.; Can, B.N.; Kodal, M.; Ozkoc, G. Compatibilization of PLA/PBAT blends by using epoxy-POSS. J. Appl. Polym. Sci. 2019, 136, 47217. [Google Scholar] [CrossRef]
- Woo, S.I.; Kim, B.O.; Jun, H.S.; Chang, H.N. Polymerization of aqueous lactic acid to prepare high molecular weight poly (lactic acid) by chain-extending with hexamethylene diisocyanate. Polym. Bull. 1995, 35, 415–421. [Google Scholar] [CrossRef]
- Tuominen, J.; Kylmä, J.; Seppälä, J. Chain extending of lactic acid oligomers. 2. increase of molecular weight with 1,6-hexamethylene diisocyanate and 2,2 0-bis(2-oxazoline). Polymer 2002, 43, 3–10. [Google Scholar] [CrossRef]
- Xiong, Z.; Zhang, L.; Ma, S.; Yang, Y.; Zhang, C.; Tang, Z.; Zhu, J. Effect of castor oil enrichment layer produced by reaction on the properties of PLA/HDI-g-starch blends. Carbohydr. Polym. 2013, 94, 235–243. [Google Scholar] [CrossRef]
- Lee, J.M.; Kim, S.H.; Jeong, H.Y.; Ahn, N.R.; Roh, H.G.; Cho, J.W.; Chun, B.C.; Oh, S.T.; Park, J.S. Preparation and characterization of polyurethane foam using a PLA/PEG polyol mixture. Fibers Polym. 2014, 15, 1349–1356. [Google Scholar] [CrossRef]
- Choi, B.; Yoo, S.; Park, S.I. Carbon footprint of packaging films made from LDPE, PLA, and PLA/PBAT blends in South Korea. Sustainability 2018, 10, 2369. [Google Scholar] [CrossRef] [Green Version]
- Supthanyakul, R.; Kaabbuathong, N.; Chirachanchai, S. Random poly(butylene succinate-co-lactic acid) as a multi-functional additive for miscibility, toughness, and clarity of PLA/PBS blends. Polymer 2016, 105, 1–9. [Google Scholar] [CrossRef]
- Arruda, L.C.; Magaton, M.; Suman Bretas, R.E.; Ueki, M.M. Influence of chain extender on mechanical, thermal and morphological properties of blown films of PLA/PBAT blends. Polym. Test 2015, 43, 27–37. [Google Scholar] [CrossRef]
- Kim, S.J.; Kwak, H.W.; Kwon, S.; Jang, H.; Park, S.I. Synthesis, characterization and properties of biodegradable poly(butylene sebacate-co-terephthalate). Polymers 2020, 12, 2389. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Xiao, Y.J.; Duan, J.; Yang, J.H.; Wang, Y.; Zhang, C.L. Accelerated hydrolytic degradation of poly(lactic acid) achieved by adding poly(butylene succinate). Polym. Bull. 2016, 73, 1067–1083. [Google Scholar] [CrossRef]
- Singla, P.; Mehta, R.; Berek, D.; Upadhyay, S.N. Microwave assisted synthesis of poly (lactic acid) and its characterization using size exclusion chromatography. J. Macromol. Sci. Part A 2012, 49, 963–970. [Google Scholar] [CrossRef]
- Setiawan, A.H.; Aulia, F. Blending of low-density polyethylene and poly-lactic acid with maleic anhydride as a compatibilizer for better environmentally food-packaging material. Int. IOP Conf. Ser. Mater. Sci. Eng. 2017, 202, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Awale, R.J.; Ali, F.B.; Azmi, A.S.; Puad, N.I.M.; Anuar, H.; Hassan, A. Enhanced flexibility of biodegradable polylactic acid/starch blends using epoxidized palm oil as plasticizer. Polymers 2018, 10, 977. [Google Scholar] [CrossRef] [Green Version]
- Chieng, B.W.; Ibrahim, N.A.; Yunus, W.M.Z.W.; Hussein, M.Z. Poly(lactic acid)/poly(ethylene glycol) polymer nanocomposites: Effects of graphene nanoplatelets. Polymers 2014, 6, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Maity, P.; Kasisomayajula, S.V.; Parameswaran, V.; Basu, S.; Gupta, N. Improvement in surface degradation properties of polymer composites due to pre-processed nanometric alumina fillers. IEEE Trans. Dielectr. Electr. Insul. 2008, 15, 63–72. [Google Scholar] [CrossRef]
- Heidarzadeh, N.; Rafizadeh, M.; Taromi, F.A.; del Valle, L.J.; Franco, L.; Puiggalí, J. Biodegradability and biocompatibility of copoly(butylene sebacate-co-terephthalate)s. Polym. Degrad. Stab. 2017, 135, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Chai, M.N.; Isa, M.I.N. The oleic acid composition effect on the carboxymethyl cellulose based biopolymer electrolyte. JCPT 2013, 3, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Nandiyanto, A.B.D.; Oktiani, R.; Ragadhita, R. How to read and interpret ftir spectroscope of organic material. Indones. J. Sci. Technol. 2019, 4, 97–118. [Google Scholar] [CrossRef]
- Maruthamuthu, S.; Nagu, M.; Natesan, M.; Palaniswamy, N. Role of air microbes on atmospheric corrosion. Curr. Sci. 2008, 94, 359–363. Available online: www.jstor.org/stable/24100344 (accessed on 25 November 2020).
- Nasar, A.S.; Libni, G. Forward and reverse reactions of N-methylaniline-blocked polyisocyanates: A clear step into double Arrhenius plots and equilibrium temperature of thermally reversible reactions. RSC Adv. 2017, 7, 34149–34159. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-J.; Min, C.-H.; Park, H.-Y.; Kim, S.-G.; Seo, K.-H. Modification of PLA/PBAT blends and thermal/mechanical properties. Appl. Chem. Eng. 2013, 24, 104–111. Available online: https://www.koreascience.or.kr/article/JAKO201319064233018.page (accessed on 3 January 2021).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specimen | PLA% (w/w) | PBSeT% (w/w) | HDI (phr) |
|---|---|---|---|
| Control | 80 | 20 | 0 |
| HDI 0.1 | 80 | 20 | 0.1 |
| HDI 0.3 | 80 | 20 | 0.3 |
| HDI 0.75 | 80 | 20 | 0.75 |
| HDI 1.0 | 80 | 20 | 1.0 |
| Unit | Control | HDI 0.1 | HDI 0.3 | HDI 0.75 | HDI 1.0 | |
|---|---|---|---|---|---|---|
| Mw | g/mol | 114,000 | 148,200 | 152,000 | 146,300 | 131,300 |
| Mn | g/mol | 45,400 | 67,800 | 57,100 | 56,800 | 54,400 |
| Đ (Dispersity) | Mw/Mn | 2.5 | 2.1 | 2.6 | 2.5 | 2.4 |
| Control | HDI 0.1 | HDI 0.3 | HDI 0.75 | HDI 1.0 | PLA | |
|---|---|---|---|---|---|---|
| Tensile strength (MPa) | 52.6 | 59.34 | 64.64 | 62.36 | 60.4 | 93.36 |
| STDV | 0.8 | 0.6 | 4.8 | 2.9 | 1.8 | 1.6 |
| Elongation at break (%) | 43.34 | 54.15 | 151.9 | 73.36 | 63.78 | 23.58 |
| STDV | 20.2 | 12.0 | 46.1 | 31.0 | 13.4 | 0.9 |
| Control | HDI 0.1 | HDI 0.3 | HDI 0.75 | HDI 1.0 | PLA | |
|---|---|---|---|---|---|---|
| Flexural Strength (MPa) | 95.3 | 106.7 | 108.7 | 108.9 | 108.9 | 151.6 |
| Flexural Modulus (GPa) | 6.5 | 7.0 | 6.4 | 6.6 | 7.1 | 11.4 |
| Yield Strength (MPa) | 81.5 | 88.3 | 91.4 | 95.0 | 95.4 | 127.5 |
| F.S. STDV | 5.7 | 3.0 | 2.6 | 4.1 | 2.7 | 4.2 |
| F.M. STDV | 0.1 | 0.1 | 0.5 | 0.5 | 0.1 | 0.8 |
| Y.S. STDV | 3.2 | 4.6 | 2.3 | 3.6 | 4.8 | 8.2 |
| Control | HDI 0.1 | HDI 0.3 | HDI 0.75 | HDI 1.0 | PLA | |
|---|---|---|---|---|---|---|
| Tm (°C) | 163.3 | 164.5 | 163.3 | 159.9/167.1 | 159.9/167.1 | 166.1 |
| ΔHf (J/g) | 21.8 | 25.2 | 34.5 | 31.6 | 32.9 | 26.0 |
| Tg (°C) | 53.4 | 54.6 | 53.9 | 58.2 | 58.0 | 59.1 |
| Tcc (°C) | 87.9 | 105.7 | 105.9 | 115.4 | 115.5 | - |
| ΔHcc (J/g) | 6.4 | 16.8 | 28.4 | 29.3 | 23.4 | - |
| Time of Degradation | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 24 h | 36 h | 48 h | 60 h | 72 h | 84 h | 96 h | 108 h | 120 h | 132 h | 144 h | ||
| Weight loss (%) | Control | 7.3 | 18.0 | 37.4 | 44.0 | 49.4 | 55.6 | 68.0 | 76.0 | 79.8 | 83.8 | 92.1 |
| HDI 0.1 | 8.0 | 14.1 | 18.4 | 24.2 | 35.7 | 42.6 | 50.3 | 55.6 | 61.4 | 64.7 | 67.5 | |
| HDI 0.3 | 10.1 | 24.9 | 27.6 | 31.9 | 38.0 | 53.4 | 58.0 | 64.1 | 69.8 | 77.6 | 85.9 | |
| HDI 0.75 | 10.6 | 18.8 | 25.6 | 29.6 | 40.2 | 51.0 | 57.2 | 62.2 | 67.6 | 73.4 | 78.6 | |
| HDI 1.0 | 9.9 | 17.8 | 28.2 | 31.9 | 38.1 | 46.4 | 51.1 | 59.8 | 65.8 | 70.5 | 71.8 | |
| PLA | 1.0 | 1.9 | 2.8 | 3.3 | 4.6 | 4.7 | 5.0 | 7.2 | 8.4 | 9.2 | 10.3 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.J.; Kwak, H.W.; Kwon, S.; Jang, H.; Park, S.-i. Characterization of PLA/PBSeT Blends Prepared with Various Hexamethylene Diisocyanate Contents. Materials 2021, 14, 197. https://doi.org/10.3390/ma14010197
Kim SJ, Kwak HW, Kwon S, Jang H, Park S-i. Characterization of PLA/PBSeT Blends Prepared with Various Hexamethylene Diisocyanate Contents. Materials. 2021; 14(1):197. https://doi.org/10.3390/ma14010197
Chicago/Turabian StyleKim, Sun Jong, Hyo Won Kwak, Sangwoo Kwon, Hyunho Jang, and Su-il Park. 2021. "Characterization of PLA/PBSeT Blends Prepared with Various Hexamethylene Diisocyanate Contents" Materials 14, no. 1: 197. https://doi.org/10.3390/ma14010197
APA StyleKim, S. J., Kwak, H. W., Kwon, S., Jang, H., & Park, S.-i. (2021). Characterization of PLA/PBSeT Blends Prepared with Various Hexamethylene Diisocyanate Contents. Materials, 14(1), 197. https://doi.org/10.3390/ma14010197