
Unusual Force Constants Guided Distortion-Triggered Loss of Long-Range Order in Phase Change Materials


Abstract
:1. Introduction
2. Materials and Methods
3. Results
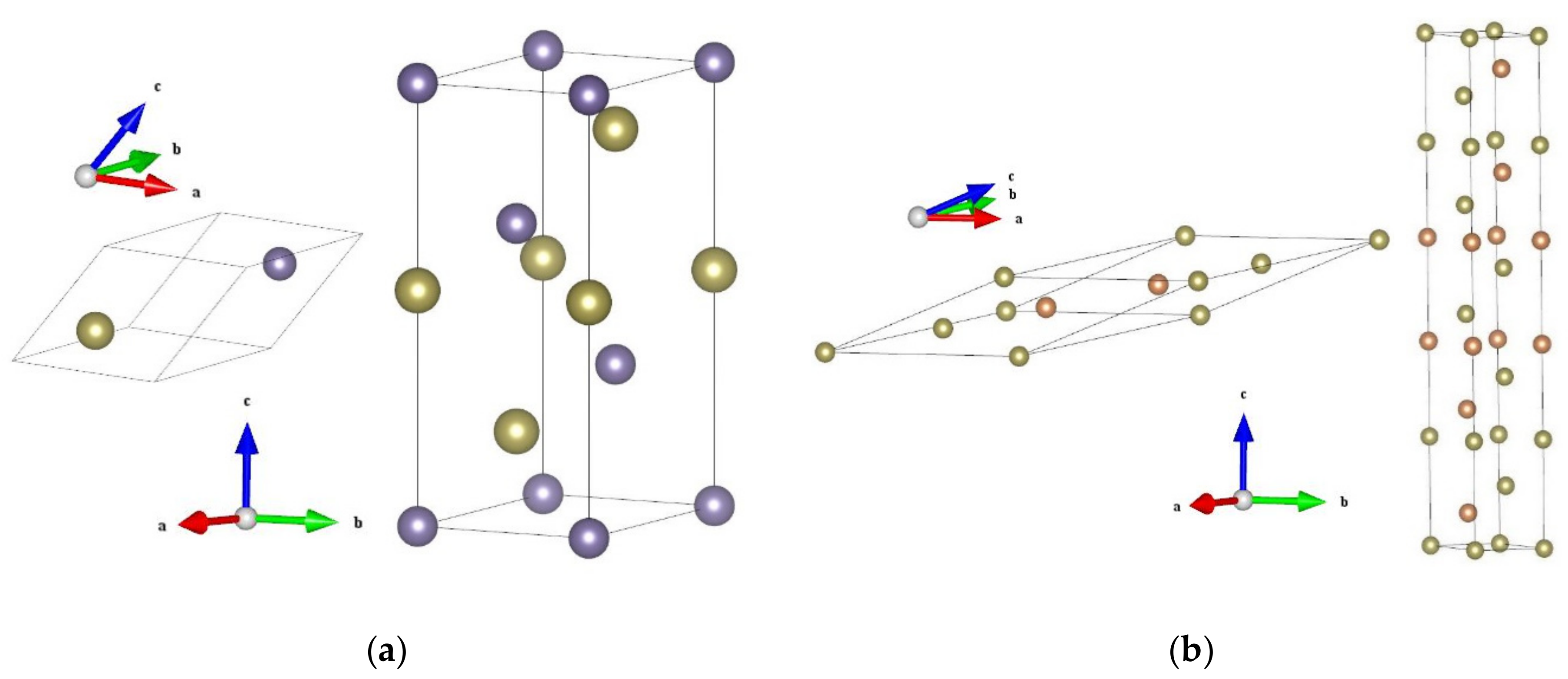
3.1. Crystalline Structures
3.2. Phonon Frequencies
3.3. Force Constants
3.4. Origin of the Unusual Force Constants
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lencer, D.; Salinga, M.; Wuttig, M. Design Rules for Phase-Change Materials in Data Storage Applications. Adv. Mater. 2011, 23, 2030–2058. [Google Scholar] [CrossRef]
- Gu, M.; Zhang, Q.; Lamon, S. Nanomaterials for optical data storage. Nat. Rev. Mater. 2016, 1, 16070. [Google Scholar] [CrossRef]
- Bilovol, V.; Fontana, M.; Rocca, J.; Chanduví, H.M.; Navarro, A.M.; Gil Rebaza, A.; Errico, L.; Liang, A.; Errandonea, D.; Ureña, A. Structural, vibrational and electronic properties in the glass-crystal transition of thin films Sb70Te30 doped with Sn. J. Alloys Compd. 2020, 845, 11. [Google Scholar] [CrossRef]
- Kolobov, A.; Krbal, M.; Fons, P.; Tominaga, J.; Uruga, T. Distortion-triggered loss of long-range order in solids with bonding energy hierarchy. Nat. Chem. 2011, 3, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Mazzarello, R.; Wuttig, M.; Ma, E. Designing crystallization in phase-change materials for universal memory and neuro-inspired computing. Nat. Rev. Mater. 2019, 4, 150–168. [Google Scholar] [CrossRef]
- Sosso, G.C.; Salvalaglio, M.; Behler, J.; Bernasconi, M.; Parrinello, M. Heterogeneous Crystallization of the Phase Change Material GeTe via Atomistic Simulations. J. Phys. Chem. C 2015, 119, 6428–6434. [Google Scholar] [CrossRef]
- Kolobov, A.V.; Fons, P.; Tominaga, J.; Hase, M. Excitation-Assisted Disordering of GeTe and Related Solids with Resonant Bonding. J. Phys. Chem. C 2014, 118, 10248–10253. [Google Scholar] [CrossRef]
- Deringer, V.; Lumeij, M.; Stoffel, R.P.; Dronskowski, R. Mechanisms of Atomic Motion Through Crystalline GeTe. Chem. Mater. 2013, 25, 2220–2226. [Google Scholar] [CrossRef]
- Krbal, M.; Kolobov, A.V.; Fons, P.; Tominaga, J.; Elliott, S.R.; Hegedus, J.; Giussani, A.; Perumal, K.; Calarco, R.; Matsunaga, T.; et al. Crystalline GeTe-based phase-change alloys: Disorder in order. Phys. Rev. B 2012, 86, 045212. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, T.; Fons, P.; Kolobov, A.V.; Tominaga, J.; Yamada, N. The order-disorder transition in GeTe: Views from different length-scales. Appl. Phys. Lett. 2011, 99, 231907. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, X.; Zhang, W.; Schäfer, L.; Reindl, J.; Bruch, F.V.; Zhou, Y.; Evang, V.; Wang, J.; Deringer, V.L.; et al. Materials Screening for Disorder-Controlled Chalcogenide Crystals for Phase-Change Memory Applications. Adv. Mater. 2021, 33, e2006221. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhou, Y.-X.; Wang, X.-D.; Chen, Y.-H.; Deringer, V.L.; Mazzarello, R.; Zhang, W. Ab initio molecular dynamics and materials design for embedded phase-change memory. NPJ Comput. Mater. 2021, 7, 29. [Google Scholar] [CrossRef]
- Kang, L.; Chen, L. Overview of the Role of Alloying Modifiers on the Performance of Phase Change Memory Materials. J. Electron. Mater. 2021, 50, 1–24. [Google Scholar] [CrossRef]
- Yuan, H.; Wang, J.; Hu, B.; Zhao, R.; Du, Y.; Zhang, S.-Y. Thermodynamic assessment of the Te-X (X = As, Si, Co) systems. Calphad 2020, 68, 101743. [Google Scholar] [CrossRef]
- Dong, C.C.; Wang, J.; Hu, B.; Zhu, L.P.; Qu, Q.; Du, Y. Thermodynamic modeling of the Te-X (X = Zr, Ce, Eu) systems. Calphad 2020, 74, 102281. [Google Scholar] [CrossRef]
- Siegrist, T.; Jost, P.; Volker, H.; Woda, M.; Merkelbach, P.; Schlockermann, C.; Wuttig, M. Disorder-induced localization in crystalline phase-change materials. Nat. Mater. 2011, 10, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Kolobov, A.V.; Fons, P.; Frenkel, A.; Ankudinov, A.L.; Tominaga, J.; Uruga, T. Understanding the phase-change mechanism of rewritable optical media. Nat. Mater. 2004, 3, 703–708. [Google Scholar] [CrossRef]
- Wuttig, M.; Yamada, N. Phase-change materials for rewriteable data storage. Nat. Mater. 2007, 6, 824–832. [Google Scholar] [CrossRef]
- Liu, X.Q.; Li, X.B.; Zhang, L.; Cheng, Y.Q.; Yan, Z.G.; Xu, M.; Han, X.D.; Zhang, S.B.; Zhang, Z.; Ma, E. New Structural Picture of the Ge2Sb2Te5 Phase-Change Alloy. Phys. Rev. Lett. 2011, 106, 025501. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Cheng, Y.Q.; Sheng, H.W.; Ma, E. Nature of Atomic Bonding and Atomic Structure in the Phase-Change Ge2Sb2Te5 Glass. Phys. Rev. Lett. 2009, 103, 195502. [Google Scholar] [CrossRef]
- Shportko, K.V.; Kremers, S.; Woda, M.; Lencer, D.; Robertson, J.; Wuttig, M. Resonant bonding in crystalline phase-change materials. Nat. Mater. 2008, 7, 653–658. [Google Scholar] [CrossRef]
- Wełnic, W.; Pamungkas, A.; Detemple, R.; Steimer, C.; Blügel, S.; Wuttig, M. Unravelling the interplay of local structure and physical properties in phase-change materials. Nat. Mater. 2006, 5, 56–62. [Google Scholar] [CrossRef]
- Kolobov, A.V.; Fons, P.; Tominaga, J. Local instability of p-type bonding makes amorphous GeTe a lone-pair semiconductor. Phys. Rev. B 2013, 87, 155204. [Google Scholar] [CrossRef]
- Cochran, W. The Relation between Phonon Frequeneies and Interatomie Force Constants. Acta Cryst. A 1971, 27, 556. [Google Scholar] [CrossRef]
- Leigh, R.S.; Szigeti, B.; Tewary, V.K. Force Constants and Lattice Frequencies. Proc. R. Soc. Lond. A 1971, 320, 505–526. [Google Scholar]
- Van de Walle, A.; Ceder, G. The effect of lattice vibrations on substitutional alloy thermodynamics. Rev. Mod. Phys. 2002, 74, 11–45. [Google Scholar] [CrossRef]
- Kong, Y.; Zhao, D.; Zhou, L.; Guo, H.; Du, Y. Energetic, mechanical, and vibrational stability of metastable OsC phase. J. Appl. Phys. 2010, 108, 083523. [Google Scholar] [CrossRef]
- Da Silva, J.L.F.; Walsh, A.; Lee, H. Insights into the structure of the stable and metastable (GeTe)m(Sb2Te3)n compounds. Phys. Rev. B 2008, 78, 224111. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Peng, L.; Li, K.; Zhu, L.; Zhou, J.; Miao, N.; Sun, Z. ALKEMIE: An intelligent computational platform for accelerating materials discovery and design. Comput. Mater. Sci. 2021, 186, 110064. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Van de Walle, A.; Asta, M.; Ceder, G. The alloy theoretic automated toolkit: A user guide. Calphad 2002, 26, 539–553. [Google Scholar] [CrossRef] [Green Version]
- Van de Walle, A. Multicomponent multisublattice alloys, nonconfigurational entropy and other additions to the Alloy Theoretic Automated Toolkit. Calphad 2009, 33, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Goldak, J.; Barrett, C.S.; Innes, D.; Youdelis, W. Structure of alpha GeTe. J. Chem. Phys. 1966, 44, 3323–3325. [Google Scholar] [CrossRef]
- Anderson, T.L.; Krause, H.B. Refinement of the Sb2Te3 and Sb2Te3Se structures and their relationship to nonstoichiometric Sb2Te3-ySey compounds. Acta Cryst. B 1974, 30, 1307–1310. [Google Scholar] [CrossRef] [Green Version]
- Shaltaf, R.; Durgun, E.; Raty, J.-Y.; Ghosez, P.; Gonze, X. Dynamical, dielectric, and elastic properties of GeTe investigated with first-principles density functional theory. Phys. Rev. B 2008, 78, 78. [Google Scholar] [CrossRef] [Green Version]
- Shaltaf, R.; Gonze, X.; Cardona, M.; Kremer, R.K.; Siegle, G. Lattice dynamics and specific heat of α-GeTe: Theoretical and experimental study. Phys. Rev. B 2009, 79, 075204. [Google Scholar] [CrossRef] [Green Version]
- Steigmeier, E.; Harbeke, G. Soft phonon mode and ferroelectricity in GeTe. Solid State Commun. 1970, 8, 1275–1279. [Google Scholar] [CrossRef]
- Richter, W.; Kohler, H.; Becker, C.R. A Raman and far-infrared investigation of phonons in the rhombohedral V2-VI3 compounds. Phys. Stat. Sol. B 1977, 84, 619–628. [Google Scholar] [CrossRef]
- Richter, W.; Krost, A.; Nowak, U.; Anastassakis, E. Anisotropy and dispersion of coupled plasmon-LO-phonon modes in Sb2Te3-need modify. J. Phys. B Condens. Matter 1982, 49, 191–198. [Google Scholar] [CrossRef]
- Sosso, G.C.; Caravati, S.; Bernasconi, M. Vibrational properties of crystalline Sb2Te3from first principles. J. Phys. Condens. Matter 2009, 21, 095410. [Google Scholar] [CrossRef]
- Antimony telluride (Sb2Te3) phonon dispersion, phonon frequencies. In Landolt-Börnstein Database: Antimony Telluride (Sb2Te3) Phonon Dispersion, Phonon Frequencies; Madelung, O.; Rössler, U.; Schulz, M. (Eds.) Landolt-Börnstein—Group III Condensed Matter; Springer: New York, NY, USA, 1983; Volume 41C. [Google Scholar]
- Rauh, H.; Geick, R.; Kohler, H.; Nucker, N.; Lehner, N. Generalized phonon density of states of the layer compounds Bi2Se3, Bi2Te3, Sb2Te3and Bi2(Te0.5Se0.5)3, (Bi0.5Sb0.5)2Te3. J. Phys. C Solid State Phys. 1981, 14, 2705–2712. [Google Scholar] [CrossRef]
- Wang, B.-T.; Souvatzis, P.; Eriksson, O.; Zhang, P. Lattice dynamics and chemical bonding in Sb2Te3 from first-principles calculations. J. Chem. Phys. 2015, 142, 6. [Google Scholar] [CrossRef]
- Andrikopoulos, K.S.; Yannopoulos, S.N.; Voyiatzis, G.A.; Kolobov, A.V.; Ribes, M.; Tominaga, J. Raman scattering study of the a-GeTe structure and possible mechanism for the amorphous to crystal transition. J. Phys. Condens. Matter 2006, 18, 965–979. [Google Scholar] [CrossRef] [Green Version]
- Drope, R. Zur Gitterdynamik von V2VI3-Verbindungen: Tunnelspektroskopische und infrarotspektroskopische Messungen an Sb2Te3-Einkristallen; RWTH Aachen University: Aachen, Germany, 1975. [Google Scholar]
- Wu, E.J.; Ceder, G.; van de Walle, A. Using bond-length-dependent transferable force constants to predict vibrational entropies in Au-Cu, Au-Pd, and Cu-Pd alloys. Phys. Rev. B 2003, 67, 7. [Google Scholar] [CrossRef] [Green Version]
- Adjaoud, O.; Steinle-Neumann, G.; Burton, B.P.; van de Walle, A. First-principles phase diagram calculations for the HfC–TiC, ZrC–TiC, and HfC–ZrC solid solutions. Phys. Rev. B 2009, 80, 7. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Z.; Ghosh, G.; van de Walle, A.; Asta, M. Transferable force-constant modeling of vibrational thermodynamic properties in fcc-basedAl−TM(TM=Ti, Zr, Hf) alloys. Phys. Rev. B 2007, 75, 15. [Google Scholar] [CrossRef]
- Kong, Y.; Li, J.H.; Kong, L.T.; Liu, B.X. Role of spatial valence charge density on the metastability of an immiscible binary metal system at equilibrium. Phys. Rev. B 2005, 72, 024209. [Google Scholar] [CrossRef]
- Caravati, S.; Bernasconi, M.; Parrinello, M. First-principles study of liquid and amorphousSb2Te3. Phys. Rev. B 2010, 81, 014201. [Google Scholar] [CrossRef]
- Sun, Z.; Zhou, J.; Blomqvist, A.; Johansson, B.; Ahuja, R. Formation of Large Voids in the Amorphous Phase-Change Memory Ge2Sb2Te5 Alloy. Phys. Rev. Lett. 2009, 102, 075504. [Google Scholar] [CrossRef] [PubMed]
- Krbal, M.; Kolobov, A.V.; Haines, J.; Fons, P.; Levelut, C.; Le Parc, R.; Hanfland, M.; Tominaga, J.; Pradel, A.; Ribes, M. Initial Structure Memory of Pressure-Induced Changes in the Phase-Change Memory Alloy Ge2Sb2Te5. Phys. Rev. Lett. 2009, 103, 115502. [Google Scholar] [CrossRef]
- Anzellini, S.; Burakovsky, L.; Turnbull, R.; Bandiello, E.; Errandonea, D. P–V–T Equation of State of Iridium Up to 80 GPa and 3100 K. Crystals 2021, 11, 452. [Google Scholar] [CrossRef]
- Jeong, K.; Park, S.; Park, D.; Ahn, M.; Han, J.; Yang, W.; Jeong, H.-S.; Cho, M.-H. Evolution of crystal structures in GeTe during phase transition. Sci. Rep. 2017, 7, 12. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | VASP | Cal. | Cal. a | Cal. b | Exp. | Exp. | Exp. |
|---|---|---|---|---|---|---|---|
| E(TO) | 90.63 | 73 | 73 | 73 | 98 | 80 | 88 |
| A1(LO) | 123.94 | 152 | 121 | 115 | 140 | 122 | 123 |
| Ref. | Present | [39] | [39] | [40] | [41] | [48] | [40] |
| Eg1 | A1g1 | Eu1 | Eu2 | A1u1 | Eg2 | A1u2 | A1g2 | Method | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 43.7 | 64.0 | 82.9 | 98.7 | 106.6 | 109.1 | 133.4 | 166.5 | VASP | Present |
| 46 | 62 | 72 | 99 | 108 | 113 | 145 | 166 | PWSCF | [44] |
| 69 | 112 | 165 | Exp. | [42] | |||||
| 43 | 56 | Exp. | [43] | ||||||
| 67 | 91 | 110 | 157 | Exp. | [49] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Cui, D.; Kong, Y.; Shen, L. Unusual Force Constants Guided Distortion-Triggered Loss of Long-Range Order in Phase Change Materials. Materials 2021, 14, 3514. https://doi.org/10.3390/ma14133514
Wang J, Cui D, Kong Y, Shen L. Unusual Force Constants Guided Distortion-Triggered Loss of Long-Range Order in Phase Change Materials. Materials. 2021; 14(13):3514. https://doi.org/10.3390/ma14133514
Chicago/Turabian StyleWang, Jiong, Dongyu Cui, Yi Kong, and Luming Shen. 2021. "Unusual Force Constants Guided Distortion-Triggered Loss of Long-Range Order in Phase Change Materials" Materials 14, no. 13: 3514. https://doi.org/10.3390/ma14133514
APA StyleWang, J., Cui, D., Kong, Y., & Shen, L. (2021). Unusual Force Constants Guided Distortion-Triggered Loss of Long-Range Order in Phase Change Materials. Materials, 14(13), 3514. https://doi.org/10.3390/ma14133514