
Effect of Silica Fume on Metakaolin Geopolymers’ Sulfuric Acid Resistance




Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Geopolymer Composition
2.3. Specimen Preparation
2.4. Exposure to Sulfuric Acid
2.5. Sample Preparation after Exposure
2.6. General Methods
2.7. SEM-EDX Mappings
3. Results
3.1. Metakaolin and Silica Fume
3.2. Unexposed Specimens
3.2.1. Compressive Strength
3.2.2. Mercury Intrusion Porosimetry
3.2.3. Fourier-Transform Infrared Spectroscopy
3.3. Sulfuric Acid
3.4. Corroded Specimens after Sulfuric Acid Exposure
3.4.1. Visual Analysis
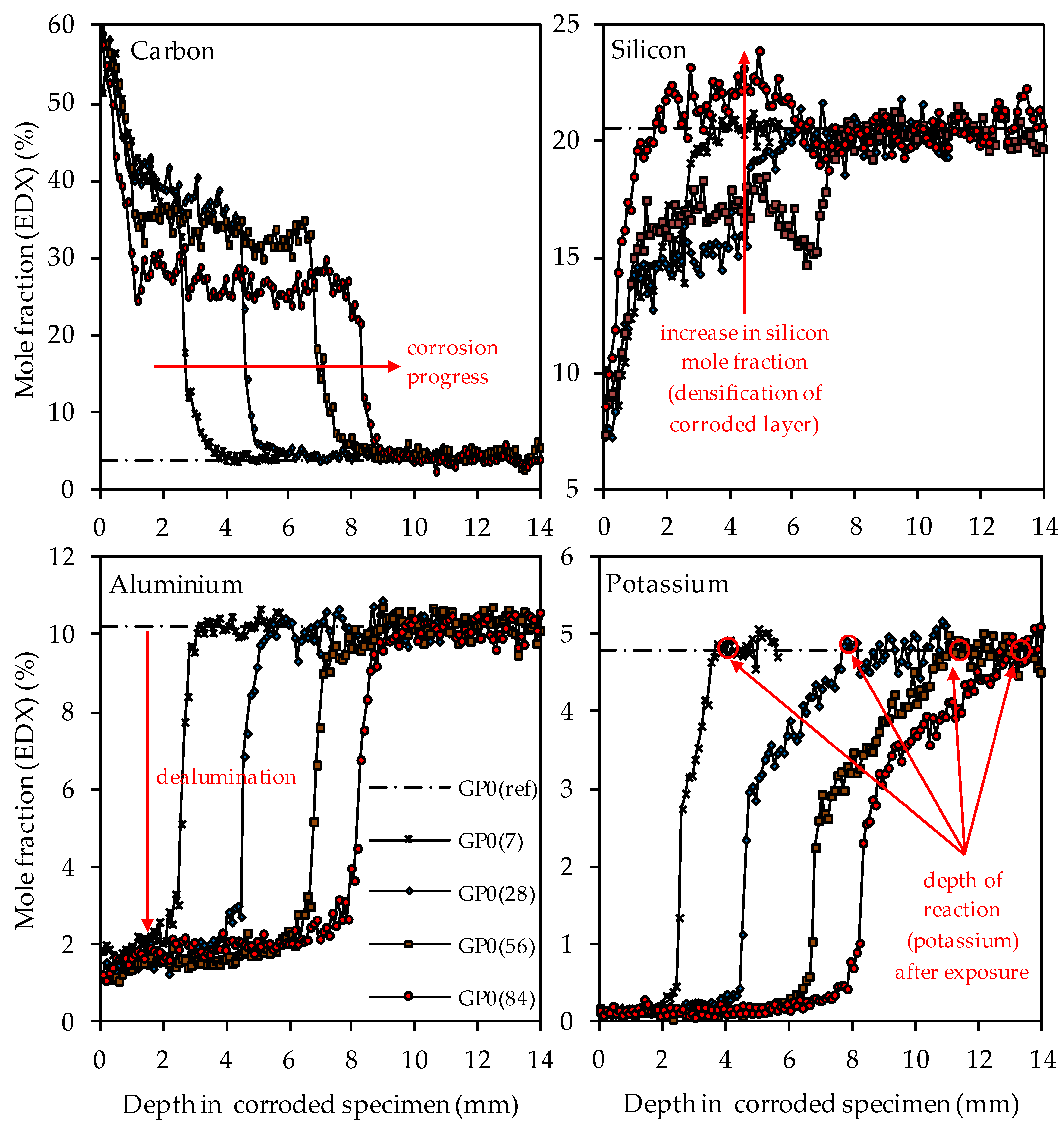
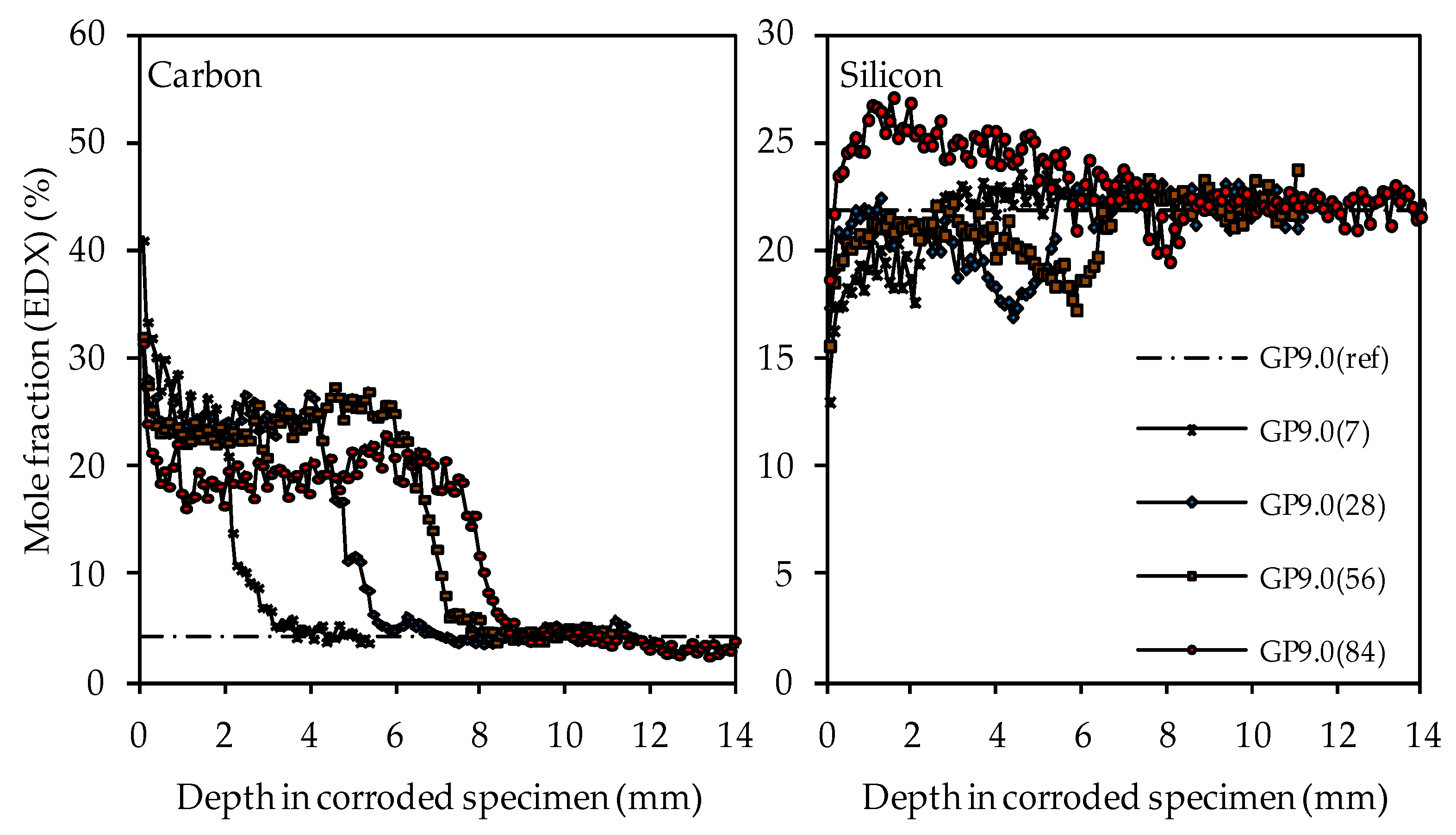
3.4.2. Depth of Erosion
3.4.3. Fourier-Transform Infrared Spectroscopy
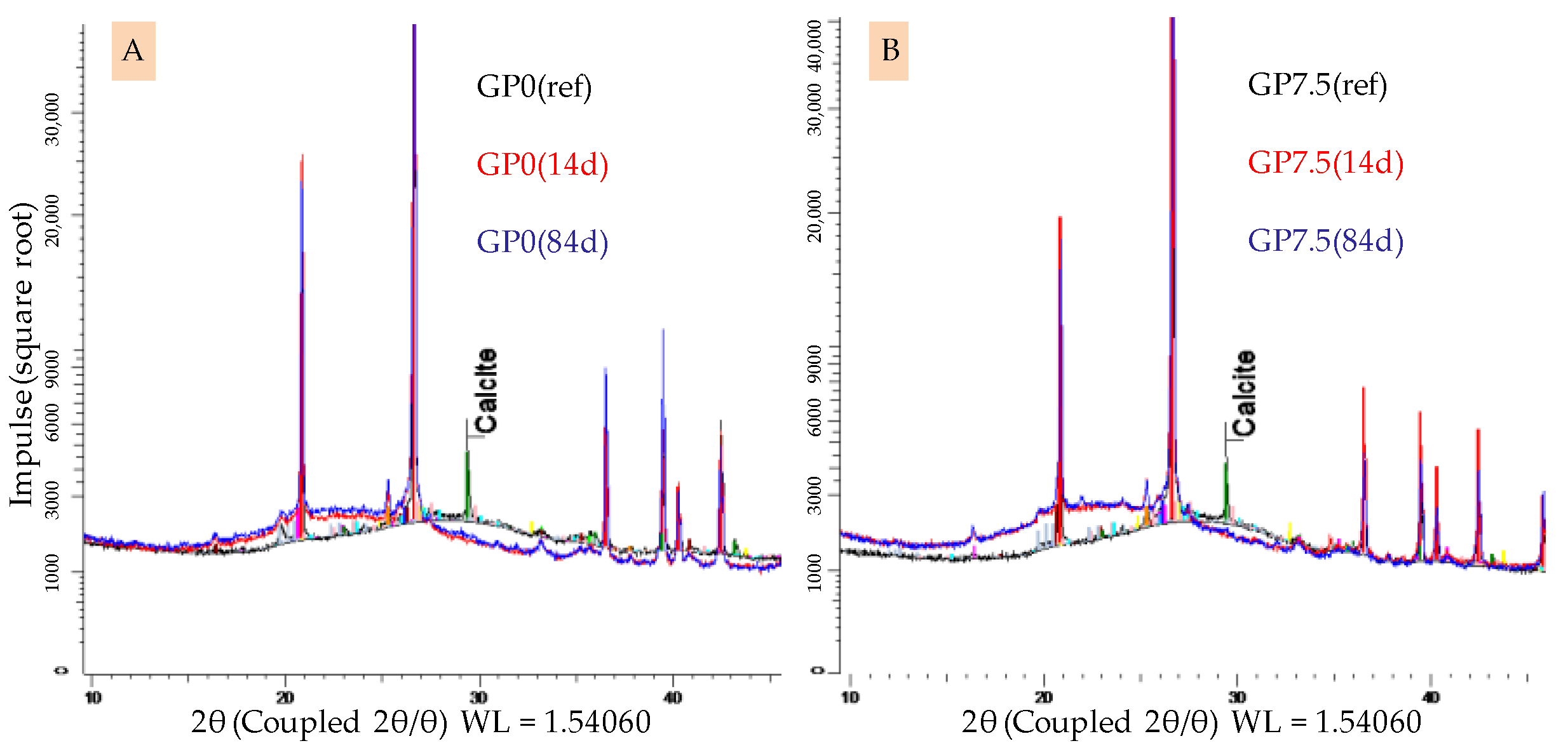
3.4.4. X-ray Diffraction
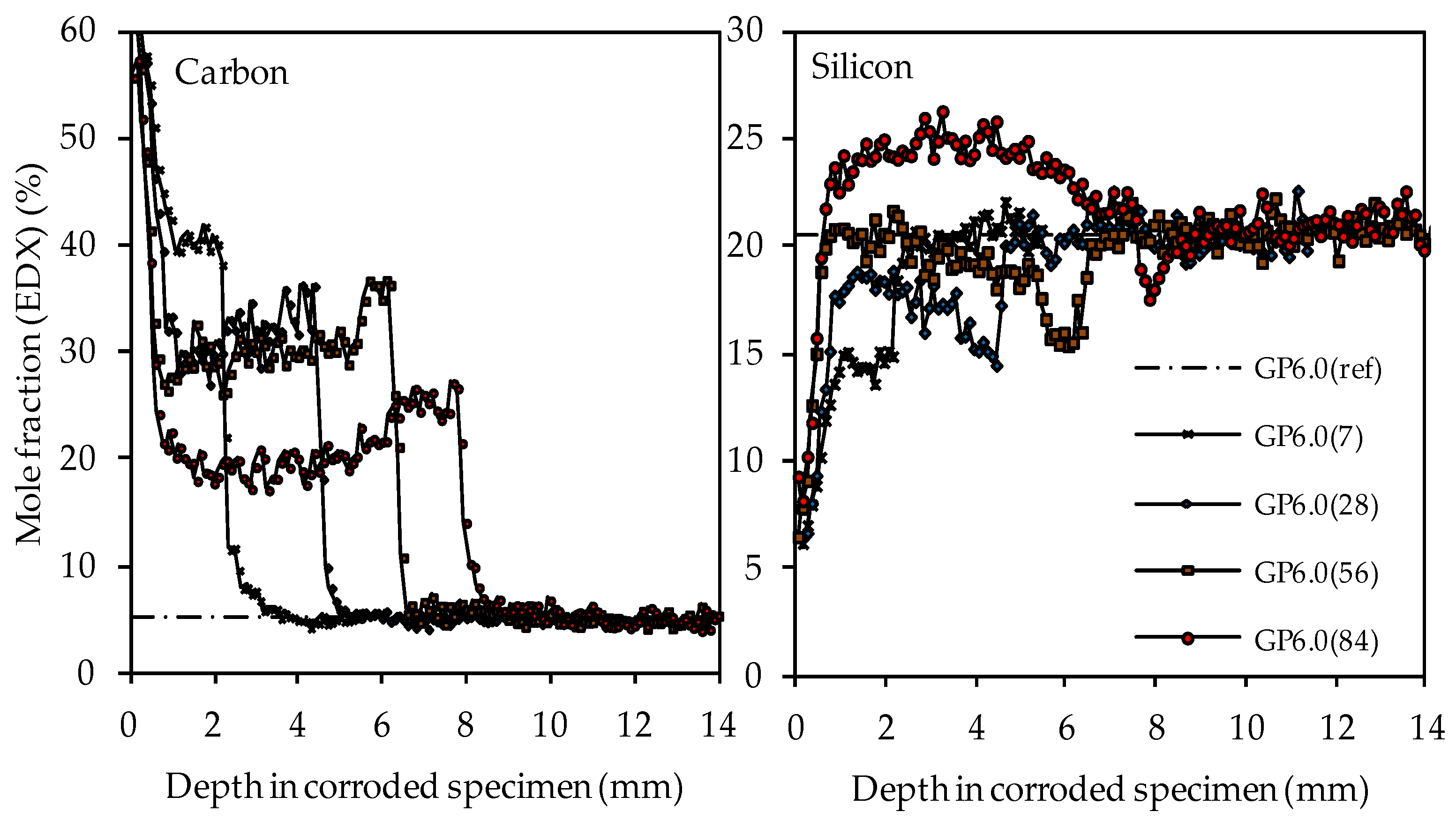
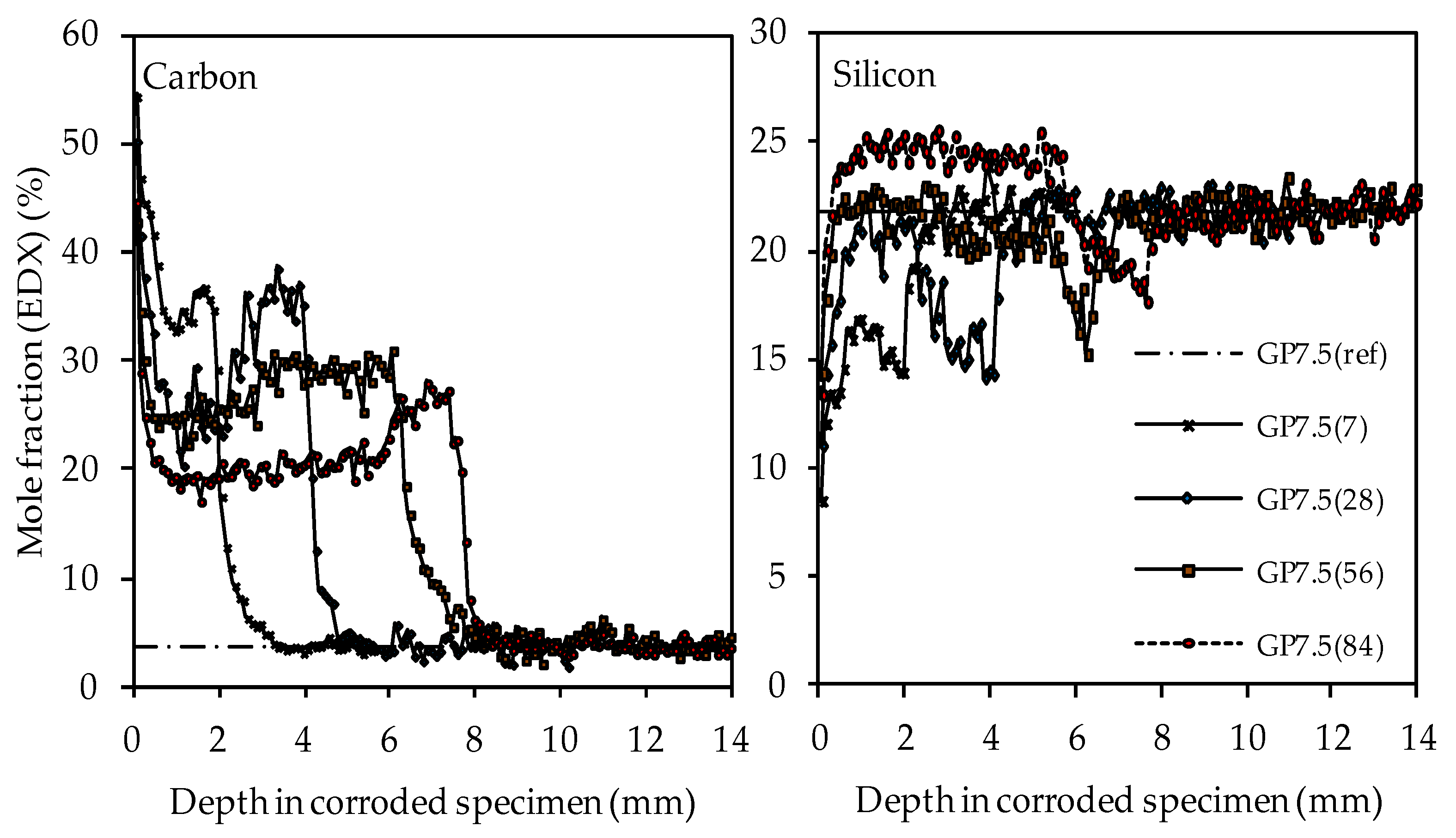
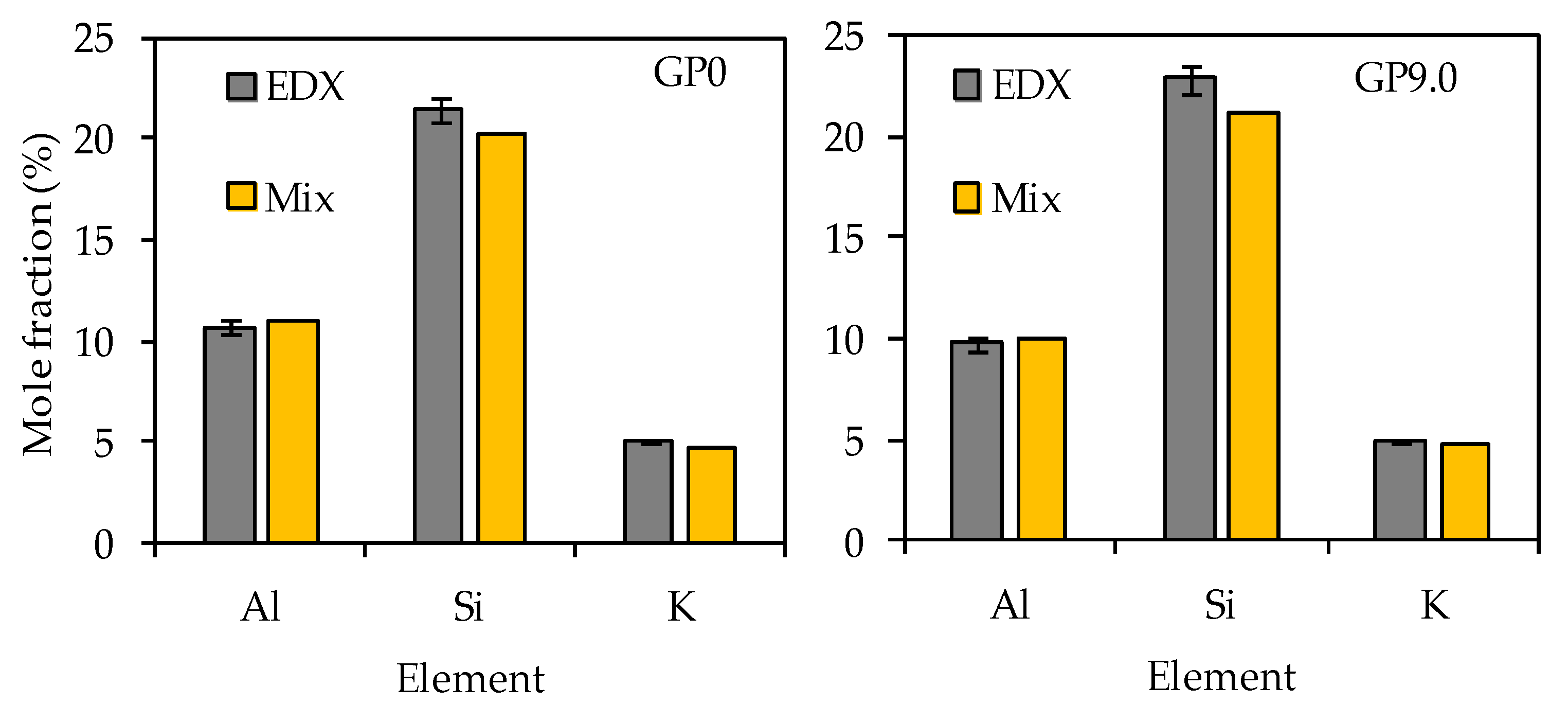
3.4.5. SEM-EDX
4. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | GP0 | GP6.0 | GP7.5 | GP9.0 |
|---|---|---|---|---|
| Si | 21.44 | 21.69 | 22.55 | 22.88 |
| Al | 10.63 | 9.74 | 9.86 | 9.75 |
| Fe | 0.81 | 0.67 | 0.76 | 0.67 |
| Ca | 0.77 | 0.65 | 0.72 | 0.65 |
| Ti | 0.18 | 0.22 | 0.17 | 0.18 |
| Na | 0.20 | 0.13 | 0.10 | 0.11 |
| K | 5.00 | 4.93 | 4.97 | 4.96 |
| Mg | 0.19 | 0.14 | 0.20 | 0.18 |
| O | 60.78 | 61.83 | 60.68 | 60.61 |
| Total | 100 | 100 | 100 | 100 |
5. Conclusions
- The substitution of 7.5% and 9.0% of metakaolin by silica fume leads to the formation of silicate-rich geopolymer structures, which are resistant against surface erosion due to dissolution reactions in sulfuric acid;
- For all of the silica fume dosages, a densification of large areas of the corroded layer of specimens can be observed due to the formation of new silicon-rich gels in the aforementioned area;
- The post-densification of large areas of the corroded layer is more pronounced and sets in at earlier exposure durations if the amount of silica fume increases, which decelerates the progress of corrosion;
- Higher proportions of silica fume have a negative influence on strength evolution of geopolymer pastes;
- The strength and durability of geopolymers could be further increased if the negative effect of higher amounts of silica fume on the geopolymerization retardation could be prevented.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koenig, A.; Dehn, F. Biogenic acid attack on concretes in biogas plants. Biosyst. Eng. 2016, 147, 226–237. [Google Scholar] [CrossRef]
- Scheydt, J.C. Mechanismen der Korrosion bei Ultrahochfestem Beton; KIT Scientific Publishing: Karlsruhe, Germany, 2014; Volume 74. [Google Scholar]
- Grengg, C.; Mittermayr, F.; Ukrainczyk, N.; Koraimann, G.; Kienesberger, S.; Dietzel, M. Advances in concrete materials for sewer systems affected by microbial induced concrete corrosion: A review. Water Res. 2018, 134, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Jiménez, A.; Palomo, A. Chemical durability of geopolymers. In Geopolymers. Structure, Processing, Properties and Industrial Applications; Provis, J.L., van Deventer, J.S.J., Eds.; Woodhead (Woodhead Publishing in Materials): Oxford, UK; Boca Raton, FL, USA, 2009; pp. 167–193. [Google Scholar]
- Lloyd, R.R.; Provis, J.L.; Deventer, J.S.J. van Pore solution composition and alkali diffusion in inorganic polymer cement. Cem. Concr. Res. 2010, 40, 1386–1392. [Google Scholar] [CrossRef]
- Ukrainczyk, N.; Muthu, M.; Vogt, O.; Koenders, E. Geopolymer, Calcium Aluminate, and Portland Cement-Based Mortars: Comparing Degradation Using Acetic Acid. Materials 2019, 12, 3115. [Google Scholar] [CrossRef] [Green Version]
- Koenig, A.; Herrmann, A.; Overmann, S.; Dehn, F. Resistance of alkali-activated binders to organic acid attack: Assessment of evaluation criteria and damage mechanisms. Constr. Build. Mater. 2017, 151, 405–413. [Google Scholar] [CrossRef]
- Bakharev, T. Resistance of geopolymer materials to acid attack. Cem. Concr. Res. 2005, 35, 658–670. [Google Scholar] [CrossRef]
- Fernández-Jiménez, A.; García-Lodeiro, I.; Palomo, A. Durability of alkali-activated fly ash cementitious materials. J. Mater. Sci. 2007, 42, 3055–3065. [Google Scholar] [CrossRef]
- Grengg, C.; Ukrainczyk, N.; Koraimann, G.; Mueller, B.; Dietzel, M.; Mittermayr, F. Long-term in situ performance of geopolymer, calcium aluminate and Portland cement-based materials exposed to microbially induced acid corrosion. Cem. Concr. Res. 2020, 131, 106034. [Google Scholar] [CrossRef]
- Okoye, F.N.; Prakash, S.; Singh, N.B. Durability of fly ash based geopolymer concrete in the presence of silica fume. J. Clean. Prod. 2017, 149, 1062–1067. [Google Scholar] [CrossRef]
- Dong, T.; Xie, S.; Wang, J.; Chen, Z.; Liu, Q. Properties and characterization of a metakaolin phosphate acid–based geopolymer synthesized in a humid environment. J. Aust. Ceram. Soc. 2020, 56, 175–184. [Google Scholar] [CrossRef]
- Zhang, B.; Guo, H.; Yuan, P.; Deng, L.; Zhong, X.; Li, Y.; Wang, Q.; Liu, D. Novel acid-based geopolymer synthesized from nanosized tubular halloysite: The role of precalcination temperature and phosphoric acid concentration. Cem. Concr. Compos. 2020, 110, 103601. [Google Scholar] [CrossRef]
- Sun, W.; Zhang, Y.; Lin, W.; Liu, Z. In situ monitoring of the hydration process of K-PS geopolymer cement with ESEM. Cem. Concr. Res. 2004, 34, 935–940. [Google Scholar] [CrossRef]
- Duxson, P.; Fernández-Jiménez, A.; Provis, J.L.; Lukey, G.C.; Palomo, A.; van Deventer, J.S. Geopolymer technology: The current state of the art. J. Mater. Sci. 2007, 42, 2917–2933. [Google Scholar] [CrossRef]
- Silva, P.D.; Sagoe-Crenstil, K.; Sirivivatnanon, V. Kinetics of geopolymerization: Role of Al2O3 and SiO2. Cem. Concr. Res. 2007, 37, 512–518. [Google Scholar] [CrossRef]
- Palomo, A.; Blanco-Varela, M.T.; Granizo, M.L.; Puertas, F.; Vazquez, T.; Grutzeck, M.W. Chemical stability of cementitious materials based on metakaolin. Cem. Concr. Res. 1999, 29, 997–1004. [Google Scholar] [CrossRef]
- Duan, P.; Yan, C.; Zhou, W.; Luo, W.; Shen, C. An investigation of the microstructure and durability of a fluidized bed fly ash–metakaolin geopolymer after heat and acid exposure. Mater. Des. 2015, 74, 125–137. [Google Scholar] [CrossRef]
- Zhuang, H.J.; Zhang, H.Y.; Xu, H. Resistance of geopolymer mortar to acid and chloride attacks. Procedia Eng. 2017, 210, 126–131. [Google Scholar] [CrossRef]
- Monique, T.T.; Julien, S.; Sylvie, R. Durability of tubular geopolymer reinforced with silica sand. New J. Glass Ceram. 2012, 2, 85–90. [Google Scholar]
- Gao, X.X.; Michaud, P.; Joussein, E.; Rossignol, S. Behavior of metakaolin-based potassium geopolymers in acidic solutions. J. Non-Cryst. Solids 2013, 380, 95–102. [Google Scholar] [CrossRef]
- Bouguermouh, K.; Bouzidi, N.; Mahtout, L.; Pérez-Villarejo, L.; Martínez-Cartas, M.L. Effect of acid attack on microstructure and composition of metakaolin-based geopolymers: The role of alkaline activator. J. Non-Cryst. Solids 2017, 463, 128–137. [Google Scholar] [CrossRef]
- Song, X.; Marosszeky, M.; Brungs, M.; Munn, R. Durability of fly ash based geopolymer concrete against sulphuric acid attack. In Proceedings of the International Conference on Durability of Building Materials and Components, Lyon, France, 17–20 April 2005; pp. 369–375. [Google Scholar]
- Thokchom, S.; Ghosh, P.; Ghosh, S. Acid resistance of fly ash based geopolymer mortars. Int. J. Recent Trends Eng. 2009, 1, 36. [Google Scholar]
- Sun, P.; Wu, H.-C. Chemical and freeze–Thaw resistance of fly ash-based inorganic mortars. Fuel 2013, 111, 740–745. [Google Scholar] [CrossRef]
- Thokchom, S. Fly ash geopolymer pastes in sulphuric acid. Int. J. Eng. Innov. Res. 2014, 3, 943–947. [Google Scholar]
- Lloyd, R.R.; Provis, J.L.; van Deventer, J.S.J. Acid resistance of inorganic polymer binders. 1. Corrosion rate. Mater. Struct. 2012, 45, 1–14. [Google Scholar] [CrossRef]
- Ariffin, M.; Bhutta, M.; Hussin, M.; Tahir, M.M.; Aziah, N. Sulfuric acid resistance of blended ash geopolymer concrete. Constr. Build. Mater. 2013, 43, 80–86. [Google Scholar] [CrossRef]
- Zhang, M.; Zhao, M.; Zhang, G.; Mann, D.; Lumsden, K.; Tao, M. Durability of red mud-fly ash based geopolymer and leaching behavior of heavy metals in sulfuric acid solutions and deionized water. Constr. Build. Mater. 2016, 124, 373–382. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Van Deventer, J.S. Microstructural characterisation of geopolymers synthesised from kaolinite/stilbite mixtures using XRD, MAS-NMR, SEM/EDX, TEM/EDX, and HREM. Cem. Concr. Res. 2002, 32, 1705–1716. [Google Scholar] [CrossRef]
- Barbosa, V.F.F.; MacKenzie, K.J.D. Synthesis and thermal behaviour of potassium sialate geopolymers. Mater. Lett. 2003, 57, 1477–1482. [Google Scholar] [CrossRef]
- Fernandez, R.; Martirena, F.; Scrivener, K.L. The origin of the pozzolanic activity of calcined clay minerals: A comparison between kaolinite, illite and montmorillonite. Cem. Concr. Res. 2011, 41, 113–122. [Google Scholar] [CrossRef]
- Vogt, O.; Ukrainczyk, N.; Ballschmiede, C.; Koenders, E. Reactivity and Microstructure of Metakaolin Based Geopolymers: Effect of Fly Ash and Liquid/Solid Contents. Materials 2019, 12, 3485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villaquirán-Caicedo, M.A.; de Gutiérrez, R.M.; Sulekar, S.; Davis, C.; Nino, J.C. Thermal properties of novel binary geopolymers based on metakaolin and alternative silica sources. Appl. Clay Sci. 2015, 118, 276–282. [Google Scholar] [CrossRef]
- Gao, K.; Lin, K.-L.; Wang, D.; Hwang, C.-L.; Tuan, B.L.A.; Shiu, H.-S.; Cheng, T.-W. Effect of nano-SiO2 on the alkali-activated characteristics of metakaolin-based geopolymers. Constr. Build. Mater. 2013, 48, 441–447. [Google Scholar] [CrossRef]
- Simonsen, M.E.; Sønderby, C.; Søgaard, E.G. Synthesis and characterization of silicate polymers. J. Sol.-Gel Sci. Technol. 2009, 50, 372. [Google Scholar] [CrossRef] [Green Version]
- Adak, D.; Sarkar, M.; Mandal, S. Structural performance of nano-silica modified fly-ash based geopolymer concrete. Constr. Build. Mater. 2017, 135, 430–439. [Google Scholar] [CrossRef]
- Jena, S.; Panigrahi, R.; Sahu, P. Mechanical and durability properties of Fly ash Geopolymer concrete with silica fume. J. Inst. Eng. India Ser. A 2019, 100, 697–705. [Google Scholar] [CrossRef]
- Ridha, S.; Abd Hamid, A.I.; Setiawan, R.A.; Shahari, A.R. Influence of sulfuric and hydrochloric acid on the resistance of geopolymer cement with nano-silica additive for oil well cement application. Int. J. Struct. Integr. 2018, 9, 616–624. [Google Scholar] [CrossRef]
- Sturm, P.; Gluth, G.; Jäger, C.; Brouwers, H.; Kühne, H.-C. Sulfuric acid resistance of one-part alkali-activated mortars. Cem. Concr. Res. 2018, 109, 54–63. [Google Scholar] [CrossRef]
- Škvára, F.; Šmilauer, V.; Hlaváček, P.; Kopeckỳ, L.; Cilova, Z. A weak alkali bond in (N, K)–A–S–H gels: Evidence from leaching and modeling. Ceram.-Silikáty 2012, 56, 374–382. [Google Scholar]
- Ukrainczyk, N. Simple Model for Alkali Leaching from Geopolymers: Effects of Raw Materials and Acetic Acid Concentration on Apparent Diffusion Coefficient. Materials 2021, 14, 1425. [Google Scholar] [CrossRef]
- Vogt, O.; Ballschmiede, C.; Ukrainczyk, N.; Koenders, E. Evaluation of Sulfuric Acid-Induced Degradation of Potassium Silicate Activated Metakaolin Geopolymers by Semi-Quantitative SEM-EDX Analysis. Materials 2020, 13, 4522. [Google Scholar] [CrossRef]
- Grengg, C.; Gluth, G.J.; Mittermayr, F.; Ukrainczyk, N.; Bertmer, M.; Buzanich, A.G.; Radtke, M.; Leis, A.; Dietzel, M. Deterioration mechanism of alkali-activated materials in sulfuric acid and the influence of Cu: A micro-to-nano structural, elemental and stable isotopic multi-proxy study. Cem. Concr. Res. 2021, 142, 106373. [Google Scholar] [CrossRef]
- Temuujin, J.; Minjigmaa, A.; Lee, M.; Chen-Tan, N.; Van Riessen, A. Characterisation of class F fly ash geopolymer pastes immersed in acid and alkaline solutions. Cem. Concr. Compos. 2011, 33, 1086–1091. [Google Scholar] [CrossRef]
- Burciaga-Díaz, O.; Escalante-García, J.I. Strength and Durability in Acid Media of Alkali Silicate-Activated Metakaolin Geopolymers. J. Am. Ceram. Soc. 2012, 95, 2307–2313. [Google Scholar] [CrossRef]
- Aiken, T.A.; Kwasny, J.; Sha, W. Performance of cementless binders produced from industrial waste products in strong acid. Clean. Eng. Technol. 2021, 2, 100035. [Google Scholar] [CrossRef]
- Aiken, T.A.; Kwasny, J.; Sha, W. Resistance of fly ash geopolymer binders to organic acids. Mater. Struct. 2020, 53, 1–18. [Google Scholar] [CrossRef]
- Ukrainczyk, N.; Vogt, O. Geopolymer leaching in water and acetic acid. RILEM Tech. Lett. 2020, 5, 163–173. [Google Scholar] [CrossRef]
- Ukrainczyk, N.; Vogt, O.; Koenders, E. Leaching of Metakaolin-Based Geopolymers in Pure Water and Acetic Acid. In Proceedings of the ICCC Prague 2019, Prague, Czech Republic, 16–20 September 2019. [Google Scholar]
- Gu, L.; Bennett, T.; Visintin, P. Sulphuric acid exposure of conventional concrete and alkali-activated concrete: Assessment of test methodologies. Constr. Build. Mater. 2019, 197, 681–692. [Google Scholar] [CrossRef]
- Islander, R.L.; Devinny, J.S.; Mansfeld, F.; Postyn, A.; Shih, H. Microbial ecology of crown corrosion in sewers. J. Environ. Eng. 1991, 117, 751–770. [Google Scholar] [CrossRef]
- Koenig, A.; Dehn, F. Main considerations for the determination and evaluation of the acid resistance of cementitious materials. Mater. Struct. 2016, 49, 1693–1703. [Google Scholar] [CrossRef]
- Kwasny, J.; Aiken, T.A.; Soutsos, M.N.; McIntosh, J.A.; Cleland, D.J. Sulfate and acid resistance of lithomarge-based geopolymer mortars. Constr. Build. Mater. 2018, 166, 537–553. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.; Visintin, P.; Bennett, T. Evaluation of accelerated degradation test methods for cementitious composites subject to sulfuric acid attack; application to conventional and alkali-activated concretes. Cem. Concr. Compos. 2018, 87, 187–204. [Google Scholar] [CrossRef]
- Aly, Z.; Vance, E.; Perera, D.; Hanna, J.; Griffith, C.; Davis, J.; Durce, D. Aqueous leachability of metakaolin-based geopolymers with molar ratios of Si/Al= 1.5–4. J. Nucl. Mater. 2008, 378, 172–179. [Google Scholar] [CrossRef]
- Buil, M.; Witier, P.; Paillere, A. Study of the Production of Hydrogen Gas by the Silica Fume in Cementitious Materials; Laboratoire Central des Ponts et Chaussees; CANMET: Paris, France, 1992. [Google Scholar]
- Delair, S.; Prud’homme, É.; Peyratout, C.; Smith, A.; Michaud, P.; Eloy, L.; Joussein, E.; Rossignol, S. Durability of inorganic foam in solution: The role of alkali elements in the geopolymer network. Corros. Sci. 2012, 59, 213–221. [Google Scholar] [CrossRef]
- Scrivener, K.; Snellings, R.; Lothenbach, B. A Practical Guide to Microstructural Analysis of Cementitious Materials; CRC Press: Boca Raton, FL, USA, 2016; Volume 540. [Google Scholar]
- Yan, S.; He, P.; Jia, D.; Duan, X.; Yang, Z.; Wang, S.; Zhou, Y. Effects of graphene oxide on the geopolymerization mechanism determined by quenching the reaction at intermediate states. RSC Adv. 2017, 7, 13498–13508. [Google Scholar] [CrossRef] [Green Version]
- Louati, S.; Baklouti, S.; Samet, B. Geopolymers based on phosphoric acid and illito-kaolinitic clay. Adv. Mater. Sci. Eng. 2016, Volume 2016, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Diercks, M.; Sand, W.; Bock, E. Microbial corrosion of concrete. Experientia 1991, 47, 514–516. [Google Scholar] [CrossRef]
- Monteny, J.; Vincke, E.; Beeldens, A.; De Belie, N.; Taerwe, L.; Van Gemert, D.; Verstraete, W. Chemical, microbiological, and in situ test methods for biogenic sulfuric acid corrosion of concrete. Cem. Concr. Res. 2000, 30, 623–634. [Google Scholar] [CrossRef]
- Deutsches Institut für Normung DIN EN. 196-1:2016-11, Prüfverfahren für Zement—Teil 1: Bestimmung der Festigkeit; Deutsche Fassung EN 196-1:2016; Beuth-Verlag: Berlin, Germany, 2016; p. 31. [Google Scholar]
- Deutsches Institut für Normung DIN EN. 12390-7:2009-07, Prüfung von Festbeton—Teil 7: Dichte von Festbeton; Deutsche Fassung EN 12390-7:2009; Beuth Publishing Din: Berlin, Germany, 2009. [Google Scholar]
- Goldstein, J.I.; Newbury, D.E.; Michael, J.R.; Ritchie, N.W.; Scott, J.H.J.; Joy, D.C. Scanning Electron Microscopy and X-Ray Microanalysis; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Caballero, L.R.; das Paiva, M.D.M.; de Fairbairn, E.M.R.; Toledo Filho, R.D. Thermal, Mechanical and Microstructural Analysis of Metakaolin Based Geopolymers. Mater. Res. 2019, 22, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Riyap, H.I.; Bewa, C.N.; Banenzoué, C.; Tchakouté, H.K.; Rüscher, C.H.; Kamseu, E.; Bignozzi, M.C.; Leonelli, C. Microstructure and mechanical, physical and structural properties of sustainable lightweight metakaolin-based geopolymer cements and mortars employing rice husk. J. Asian Ceram. Soc. 2019, 7, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Li, T.; Li, X.; Chou, K.-C.; Hou, X. Morphological evolution of low-grade silica fume at elevated temperature. High. Temp. Mater. Process. 2017, 36, 607–613. [Google Scholar] [CrossRef]
- Zanoletti, A.; Vassura, I.; Venturini, E.; Monai, M.; Montini, T.; Federici, S.; Zacco, A.; Treccani, L.; Bontempi, E. A new porous hybrid material derived from silica fume and alginate for sustainable pollutants reduction. Front. Chem. 2018, 6, 60. [Google Scholar] [CrossRef]
- Zhong, Y.; Qiu, X.; Gao, J.; Guo, Z. Chemical Structure of Si–O in Silica Fume from Ferrosilicon Production and Its Reactivity in Alkali Dissolution. ISIJ Int. 2019, 59, 1098–1104. [Google Scholar] [CrossRef] [Green Version]
- El Alouani, M.; Alehyen, S.; El Achouri, M. Preparation, characterization, and application of Metakaolin-based geopolymer for removal of methylene blue from aqueous solution. J. Chem. 2019, 2019, 1–14. [Google Scholar] [CrossRef]
- Criado, M.; Palomo, A.; Fernández-Jiménez, A. Alkali activation of fly ashes. Part 1: Effect of curing conditions on the carbonation of the reaction products. Fuel 2005, 84, 2048–2054. [Google Scholar] [CrossRef]
- Kouamo, H.T.; Elimbi, A.; Mbey, J.A.; Sabouang, C.J.N.; Njopwouo, D. The effect of adding alumina-oxide to metakaolin and volcanic ash on geopolymer products: A comparative study. Constr. Build. Mater. 2012, 35, 960–969. [Google Scholar] [CrossRef]
- Ren, D.; Yan, C.; Duan, P.; Zhang, Z.; Li, L.; Yan, Z. Durability performances of wollastonite, tremolite and basalt fiber-reinforced metakaolin geopolymer composites under sulfate and chloride attack. Constr. Build. Mater. 2017, 134, 56–66. [Google Scholar] [CrossRef]
- Hajimohammadi, A.; Provis, J.L.; van Deventer, J.S.J. The effect of silica availability on the mechanism of geopolymerisation. Cem. Concr. Res. 2011, 41, 210–216. [Google Scholar] [CrossRef]
- Granizo, M.L.; Blanco-Varela, M.T.; Martínez-Ramírez, S. Alkali activation of metakaolins: Parameters affecting mechanical, structural and microstructural properties. J. Mater. Sci. 2007, 42, 2934–2943. [Google Scholar] [CrossRef]
- Kenne Diffo, B.; Elimbi, A.; Cyr, M.; Dika Manga, J.; Tchakoute Kouamo, H. Effect of the rate of calcination of kaolin on the properties of metakaolin-based geopolymers. J. Asian Ceram. Soc. 2015, 3, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Aly, Z.; Vance, E.; Perera, D. Aqueous dissolution of sodium aluminosilicate geopolymers derived from metakaolin. J. Nucl. Mater. 2012, 424, 164–170. [Google Scholar] [CrossRef]
- Lee, W.; Van Deventer, J. Structural reorganisation of class F fly ash in alkaline silicate solutions. Colloids Surf. Physicochem. Eng. Asp. 2002, 211, 49–66. [Google Scholar] [CrossRef]
- Baščarević, Z.; Komljenović, M.; Miladinović, Z.; Nikolić, V.; Marjanović, N.; Žujović, Z.; Petrović, R. Effects of the concentrated NH4NO3 solution on mechanical properties and structure of the fly ash based geopolymers. Constr. Build. Mater. 2013, 41, 570–579. [Google Scholar] [CrossRef]
- Duxson, P.; Provis, J.L.; Lukey, G.C.; Mallicoat, S.W.; Kriven, W.M.; van Deventer, J.S.J. Understanding the relationship between geopolymer composition, microstructure and mechanical properties. Colloids Surf. Physicochem. Eng. Asp. 2005, 269, 47–58. [Google Scholar] [CrossRef]
- Criado, M.; Fernández-Jiménez, A.; Palomo, A.; Sobrados, I.; Sanz, J. Effect of the SiO2/Na2O ratio on the alkali activation of fly ash. Part II: 29Si MAS-NMR Survey. Microporous Mesoporous Mater. 2008, 109, 525–534. [Google Scholar] [CrossRef]
- Alanazi, H.; Yang, M.; Zhang, D.; Gao, Z. (Jerry). Bond strength of PCC pavement repairs using metakaolin-based geopolymer mortar. Cem. Concr. Compos. 2016, 65, 75–82. [Google Scholar] [CrossRef]
- Svensson, I.L.; Sjöberg, S.; Öhman, L.-O. Polysilicate equilibria in concentrated sodium silicate solutions. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1986, 82, 3635–3646. [Google Scholar] [CrossRef]
- Engelhardt, L.; Zeigan, D.; Jancke, H.; Wieker, W.; Hoebbel, D. 29Si-NMR-Spektroskopie an Silicatlösungen. II. Zur Abhängigkeit der Struktur der Silicatanionen in wä\s srigen Natriumsilicatlösungen vom Na: Si-Verhältnis. Z. Für Anorg. Allg. Chem. 1975, 418, 17–28. [Google Scholar] [CrossRef]
- McCormick, A.V.; Bell, A. The solution chemistry of zeolite precursors. Catal. Rev. Eng. 1989, 31, 97–127. [Google Scholar] [CrossRef]
- McCormick, A.; Bell, A.; Radke, C. Multinuclear NMR investigation of the formation of aluminosilicate anions. J. Phys. Chem. 1989, 93, 1741–1744. [Google Scholar] [CrossRef]
- Garcia-Lodeiro, I.; Palomo, A.; Fernández-Jiménez, A. Crucial insights on the mix design of alkali-activated cement-based binders. In Handbook of Alkali-Activated Cements, Mortars and Concretes; Elsevier: Amsterdam, The Netherlands, 2015; pp. 49–73. [Google Scholar]
- De Jong, B.; Brown, G.E., Jr. Polymerization of silicate and aluminate tetrahedra in glasses, melts, and aqueous solutions—I. Electronic structure of H6Si2O7, H6AlSiO71-, and H6Al2O72-. Geochim. Cosmochim. Acta 1980, 44, 491–511. [Google Scholar] [CrossRef]
- Duxson, P.; Mallicoat, S.; Lukey, G.C.; Kriven, W.M.; van Deventer, J.S. Microstructural characterisation of metakaolin-based geopolymers. Adv. Ceram. Matrix Compos. X 2006, 165, 71–85. [Google Scholar]
- Duxson, P.; Lukey, G.; Separovic, F.; Van Deventer, J. Effect of alkali cations on aluminum incorporation in geopolymeric gels. Ind. Eng. Chem. Res. 2005, 44, 832–839. [Google Scholar] [CrossRef]



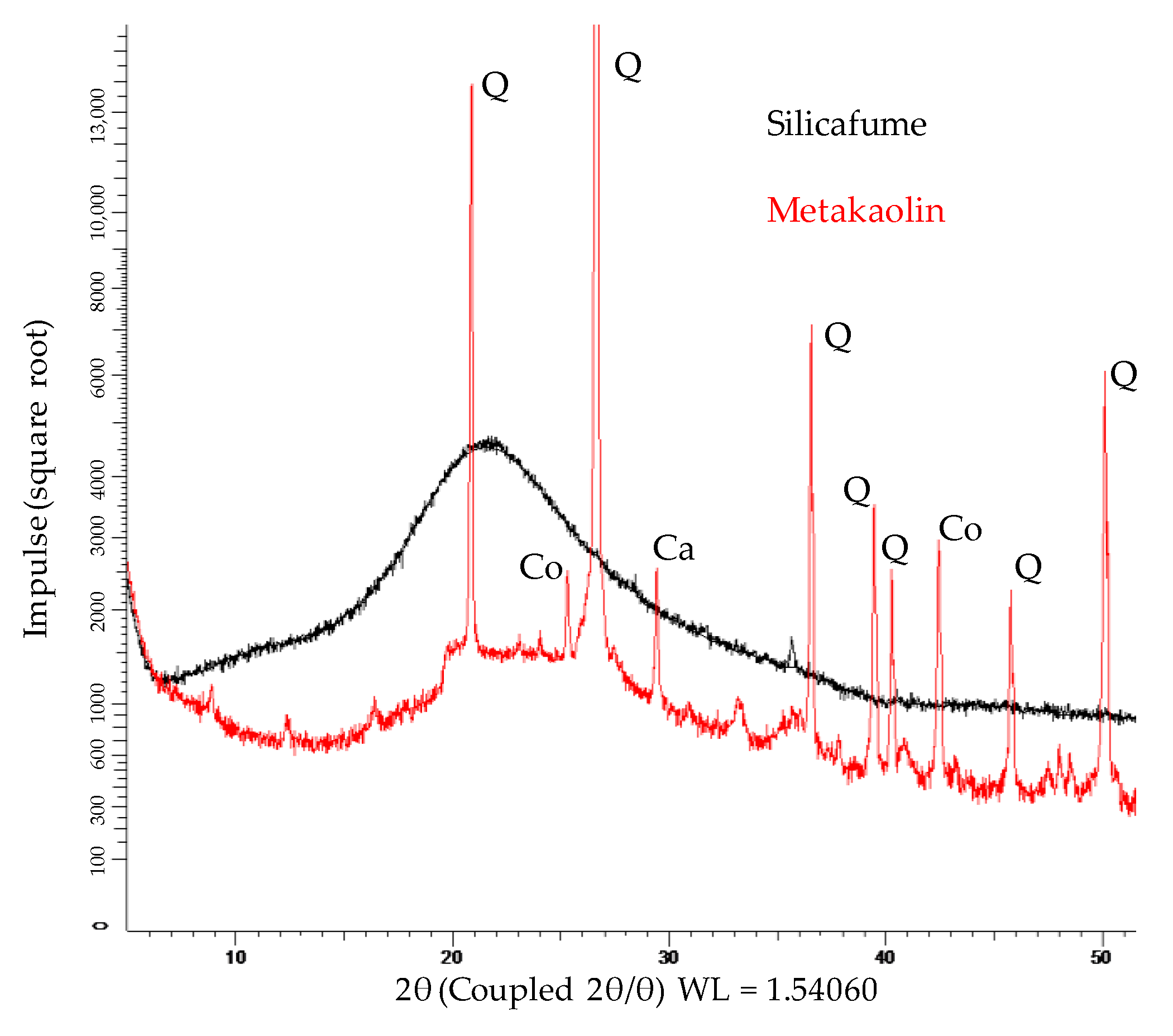
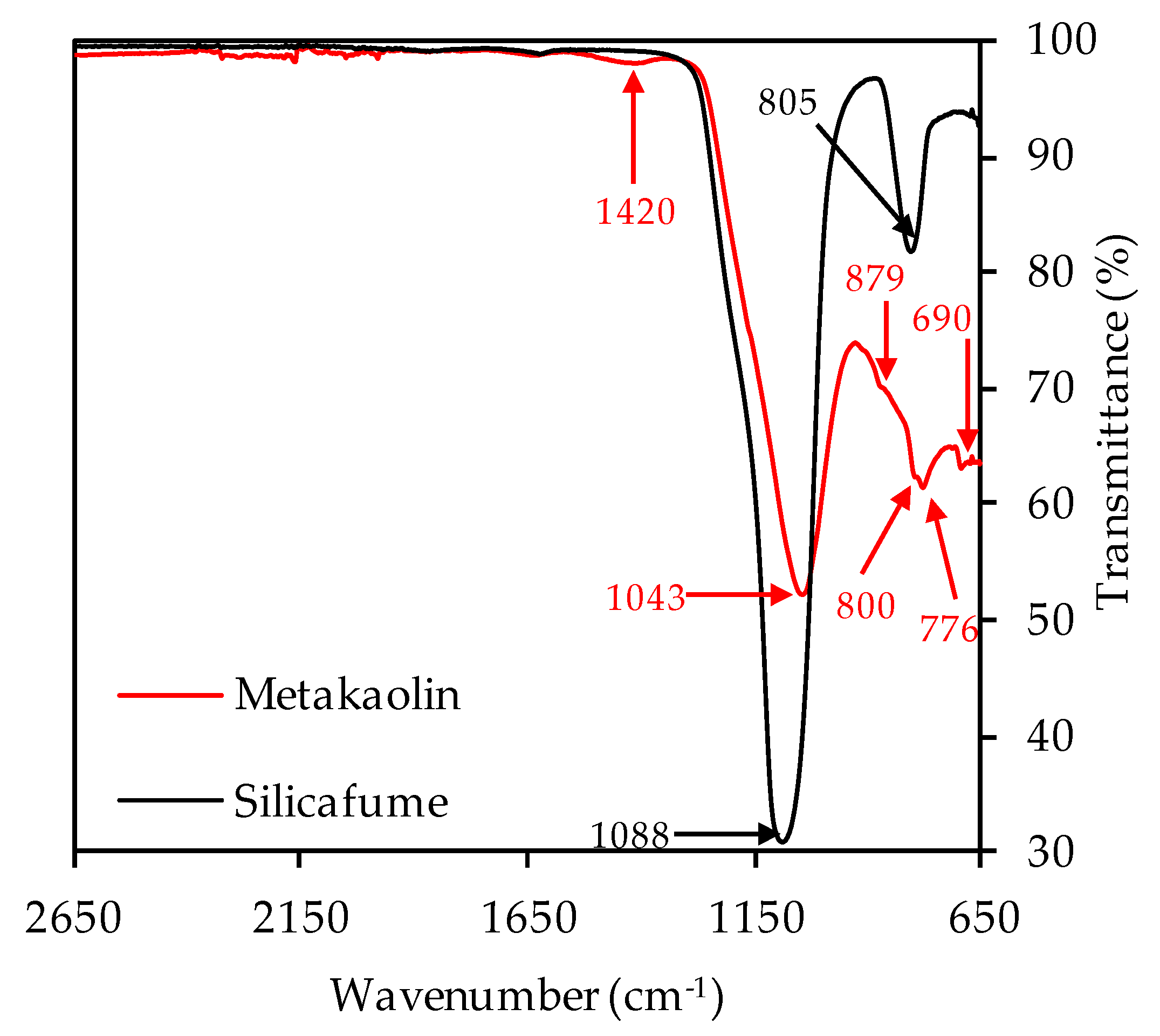

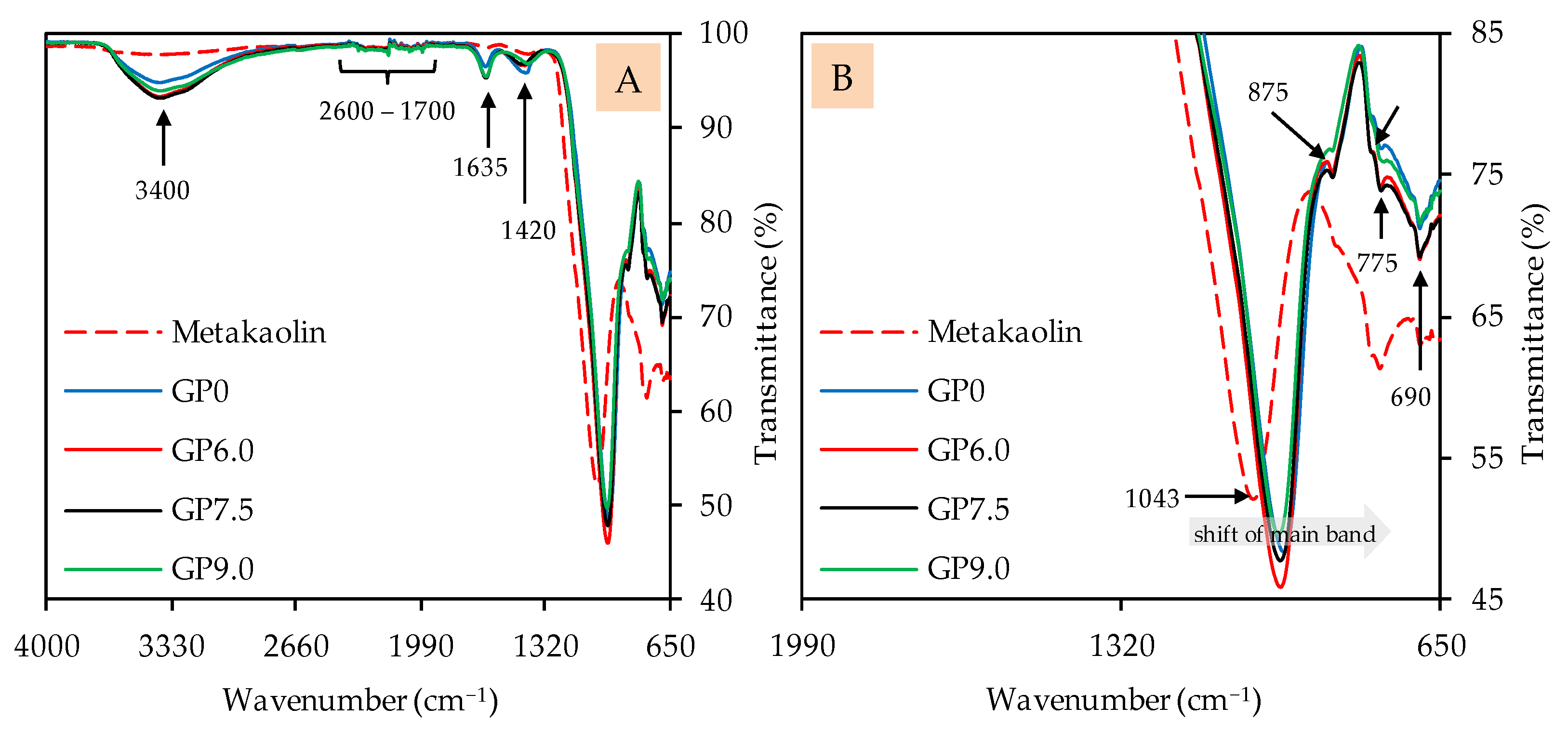



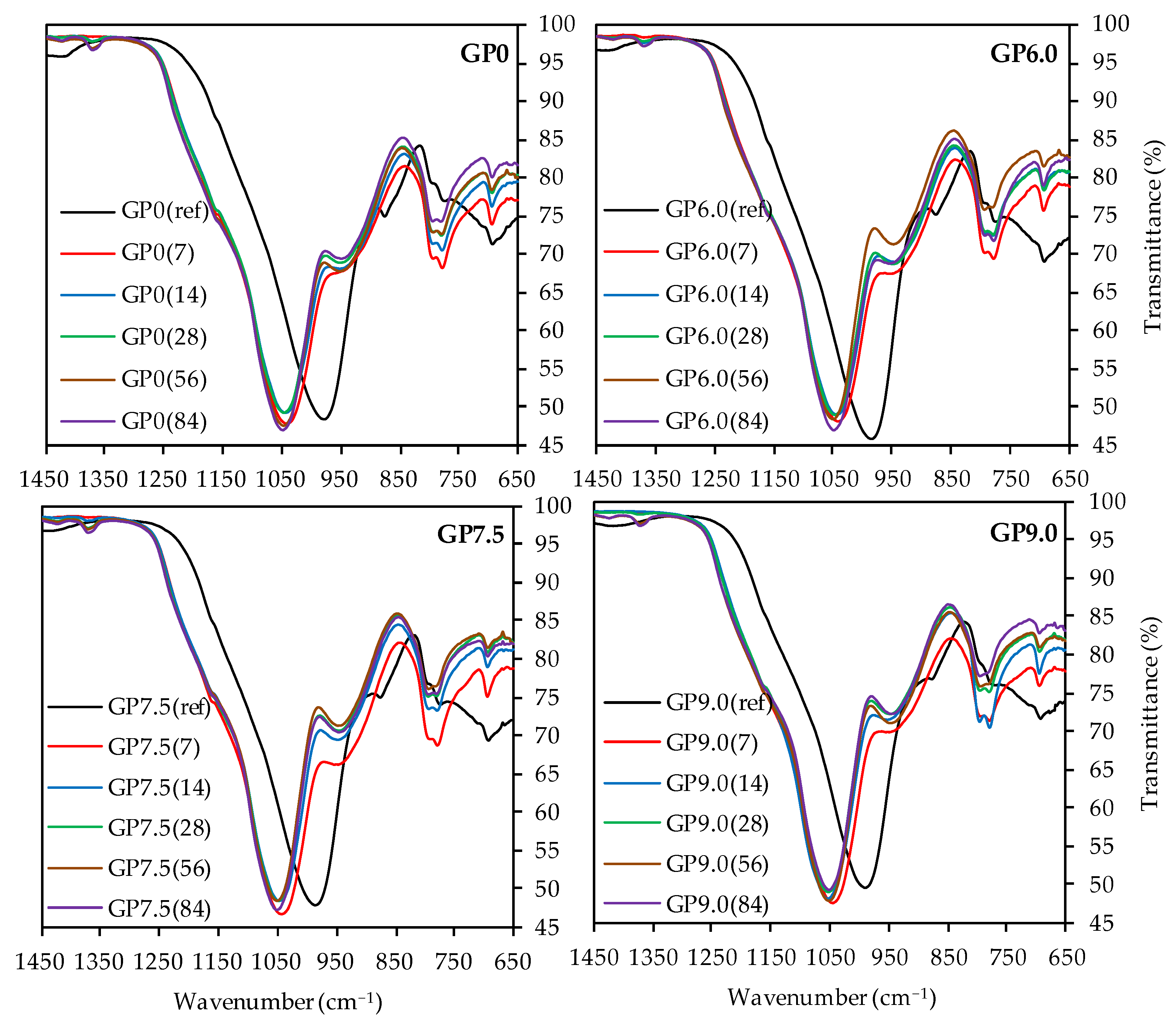
| Oxides | Metakaolin (wt%) | Silica Fume (wt%) |
|---|---|---|
| SiO2 | 58.80 | 97.02 |
| Al2O3 | 32.79 | 0.55 |
| Fe2O3 | 3.50 | 0.21 |
| CaO | 2.35 | 0.32 |
| TiO2 | 1.80 | 0.00 |
| Na2O | 0.06 | 0.18 |
| K2O | 0.35 | 0.98 |
| MgO | 0.17 | 0.49 |
| Geopolymer | Metakaolin (%) | Silica Fume (%) | Potassium Silicate Solution (%) | l/s (-) |
|---|---|---|---|---|
| GP0 | 64.81 | 0.00 | 35.19 | 0.54 |
| GP6.0 | 60.92 | 3.89 | 35.19 | 0.54 |
| GP7.5 | 59.95 | 4.86 | 35.19 | 0.54 |
| GP9.0 | 58.98 | 5.83 | 35.19 | 0.54 |
| Element | GP0 | GP6.0 | GP7.5 | GP9.0 |
|---|---|---|---|---|
| Si | 20.22 | 20.85 | 21.01 | 21.17 |
| Al | 11.05 | 10.38 | 10.22 | 10.05 |
| Fe | 0.75 | 0.71 | 0.70 | 0.69 |
| Ca | 0.72 | 0.68 | 0.67 | 0.66 |
| Ti | 0.39 | 0.36 | 0.36 | 0.35 |
| Na | 0.03 | 0.04 | 0.04 | 0.04 |
| K | 4.68 | 4.69 | 4.69 | 4.69 |
| Mg | 0.07 | 0.08 | 0.08 | 0.08 |
| O | 62.08 | 62.20 | 62.23 | 62.26 |
| Total | 100 | 100 | 100 | 100 |
| Geopolymer | Compressive Strength (MPa) | ||
|---|---|---|---|
| 1d | 7d | 28d | |
| GP0 | 60.8 | 53.6 | 58.0 |
| GP6.0 | 13.9 | 65.7 | 81.3 |
| GP7.5 | - | 15.4 | 71.9 |
| GP9.0 | - | - | 10.7 |
| Geopolymer | Depth of Erosion (mm) | ||||
|---|---|---|---|---|---|
| 7d | 14d | 28d | 56d | 84d | |
| GP0 | 1.04 | 1.01 | 1.14 | 1.16 | 1.07 |
| GP6.0 | 0.89 | 0.78 | 0.96 | 0.83 | 0.94 |
| GP7.5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| GP9.0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vogt, O.; Ukrainczyk, N.; Koenders, E. Effect of Silica Fume on Metakaolin Geopolymers’ Sulfuric Acid Resistance. Materials 2021, 14, 5396. https://doi.org/10.3390/ma14185396
Vogt O, Ukrainczyk N, Koenders E. Effect of Silica Fume on Metakaolin Geopolymers’ Sulfuric Acid Resistance. Materials. 2021; 14(18):5396. https://doi.org/10.3390/ma14185396
Chicago/Turabian StyleVogt, Oliver, Neven Ukrainczyk, and Eddie Koenders. 2021. "Effect of Silica Fume on Metakaolin Geopolymers’ Sulfuric Acid Resistance" Materials 14, no. 18: 5396. https://doi.org/10.3390/ma14185396
APA StyleVogt, O., Ukrainczyk, N., & Koenders, E. (2021). Effect of Silica Fume on Metakaolin Geopolymers’ Sulfuric Acid Resistance. Materials, 14(18), 5396. https://doi.org/10.3390/ma14185396