Porous Materials Based on Poly(methylvinylsiloxane) Cross-Linked with 1,3,5,7-Tetramethylcyclotetrasiloxane in High Internal Phase Emulsion as Precursors to Si-C-O and Si-C-O/Pd Ceramics


Abstract
:1. Introduction
2. Materials and Methods
2.1. Starting Materials
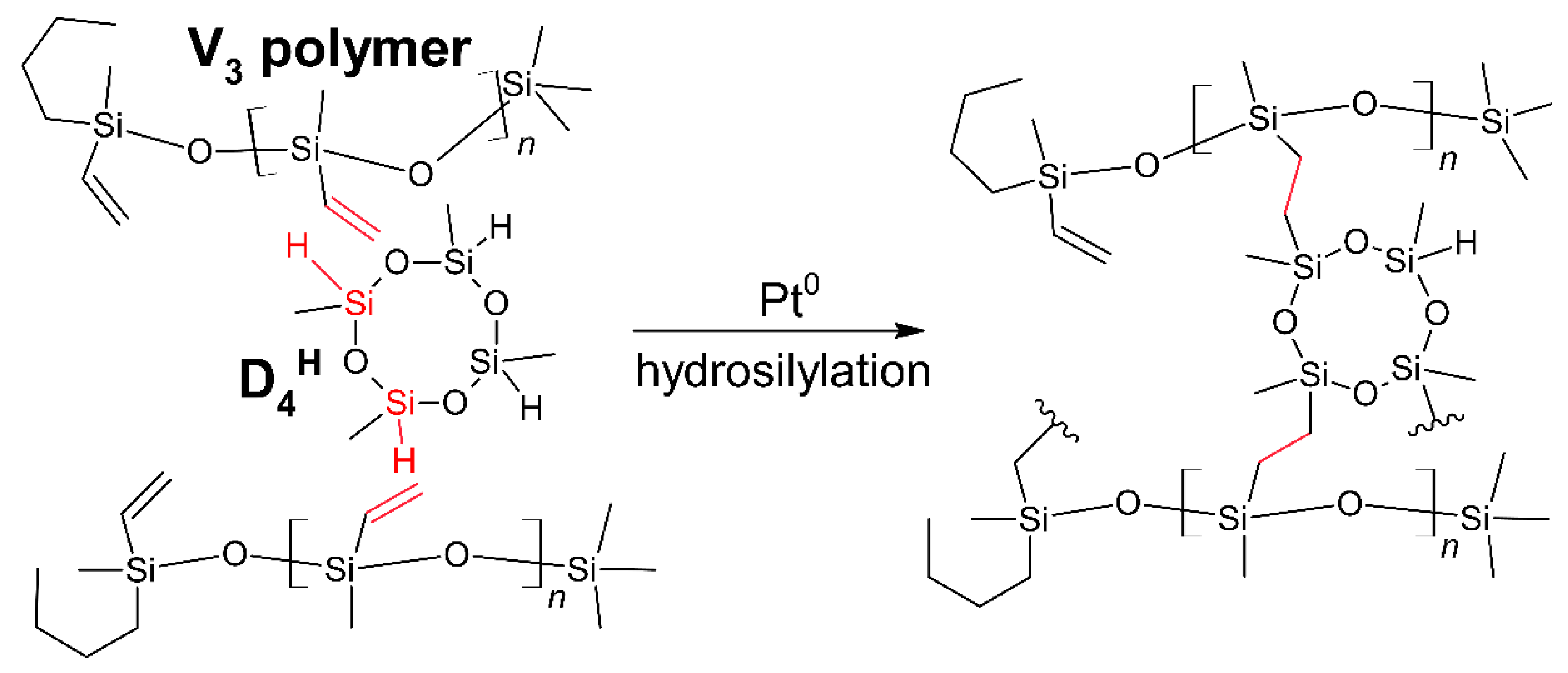
2.2. Preparation of Polysiloxane, Polysiloxane-Pd Networks, and the Pyrolyzed Materials
2.3. Characterization Methods
- D—mean crystallite size (nm)
- K—Scherrer constant; in the performed calculations K = 1was applied
- λ—wavelength of the X ray radiation, λ = 0.15406 nm for CuKα lamp
- β—full width at half maximum (FWHM) of the Pd(100) peak (deg)
- θ—Bragg diffraction angle (deg)
3. Results and Discussion
3.1. V3 Polymer-Based PolyHIPEs before and after Pyrolysis
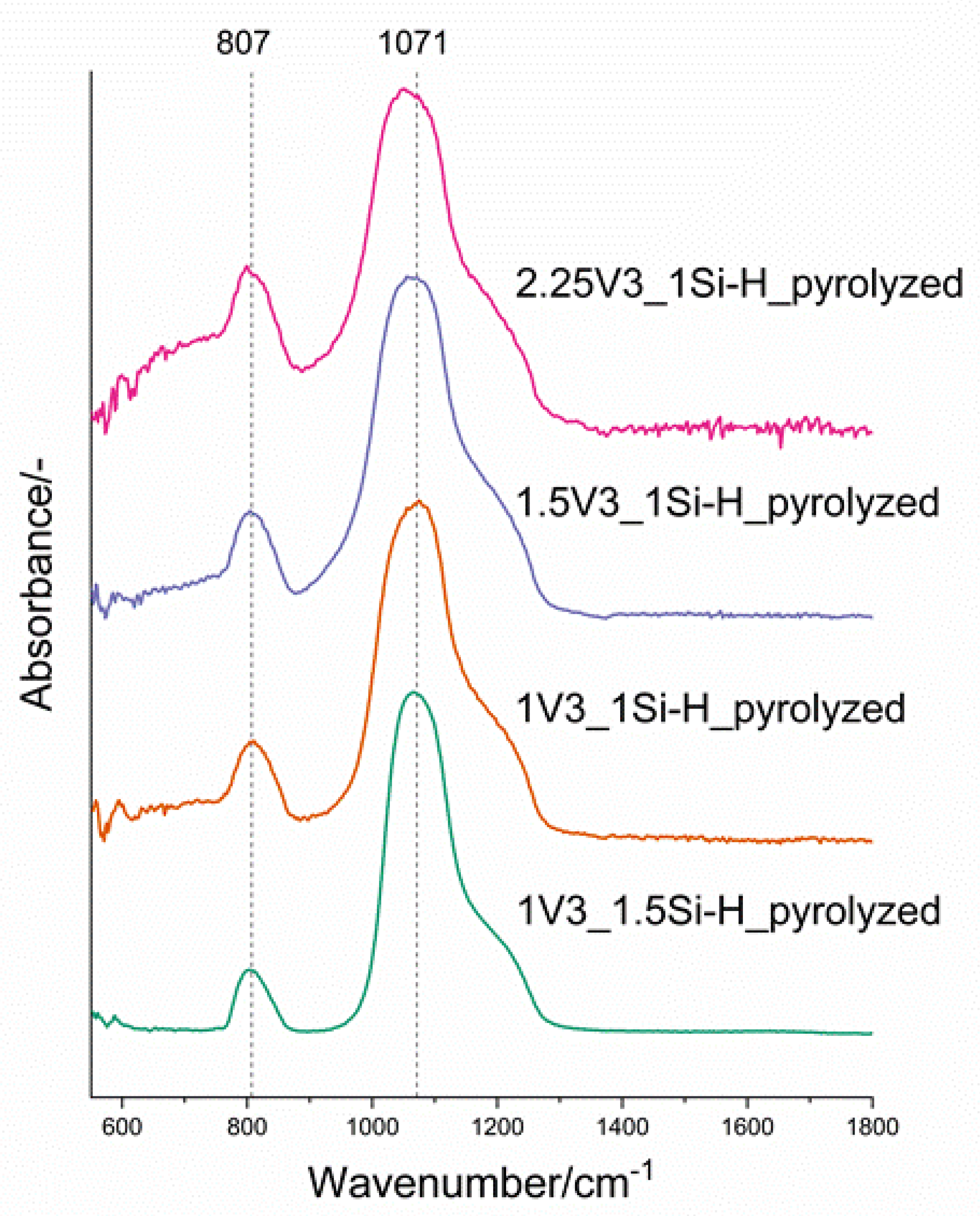
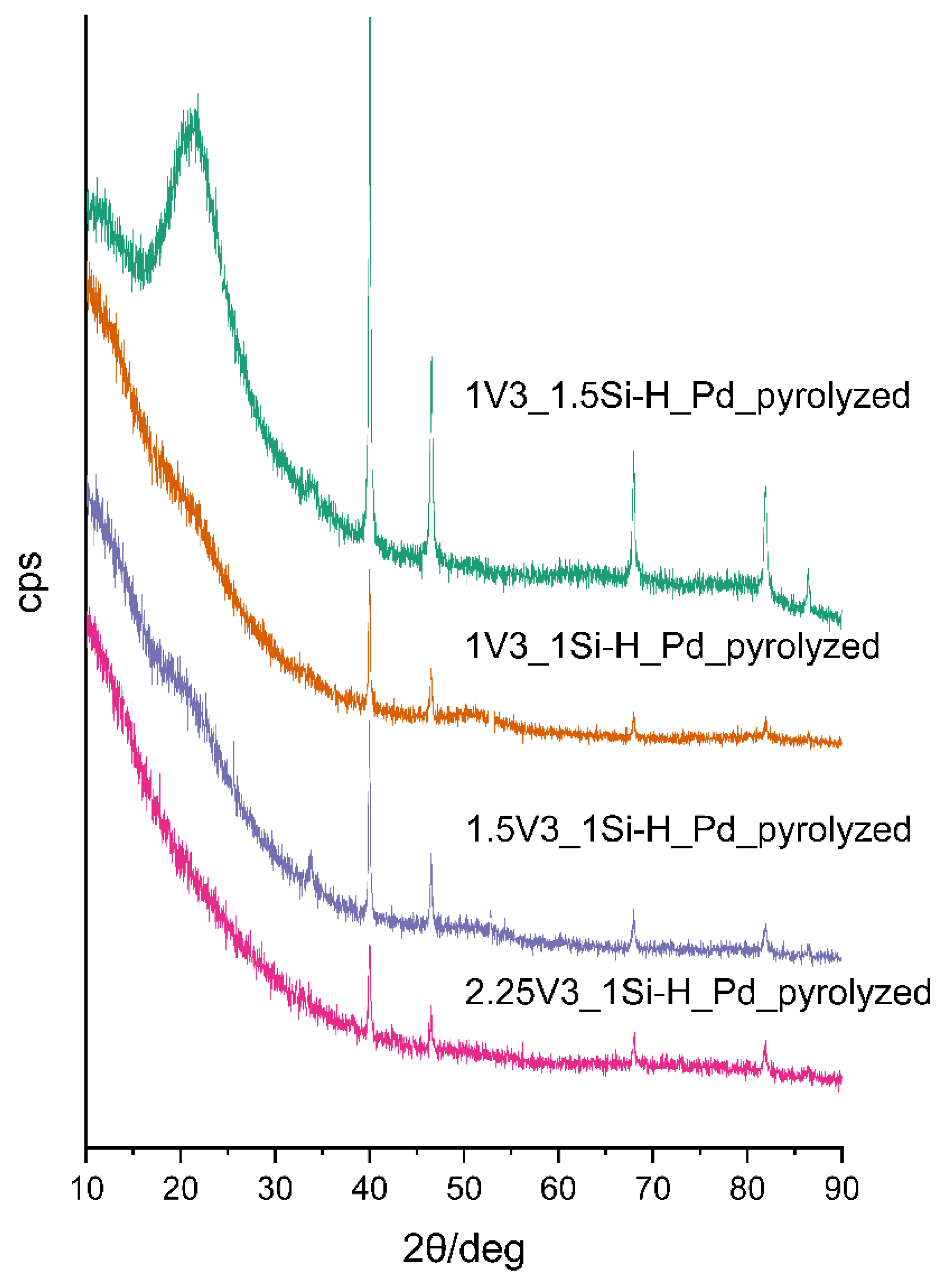
3.2. Pd-Containing Materials: XRD, SEM, and Spectroscopic Studies
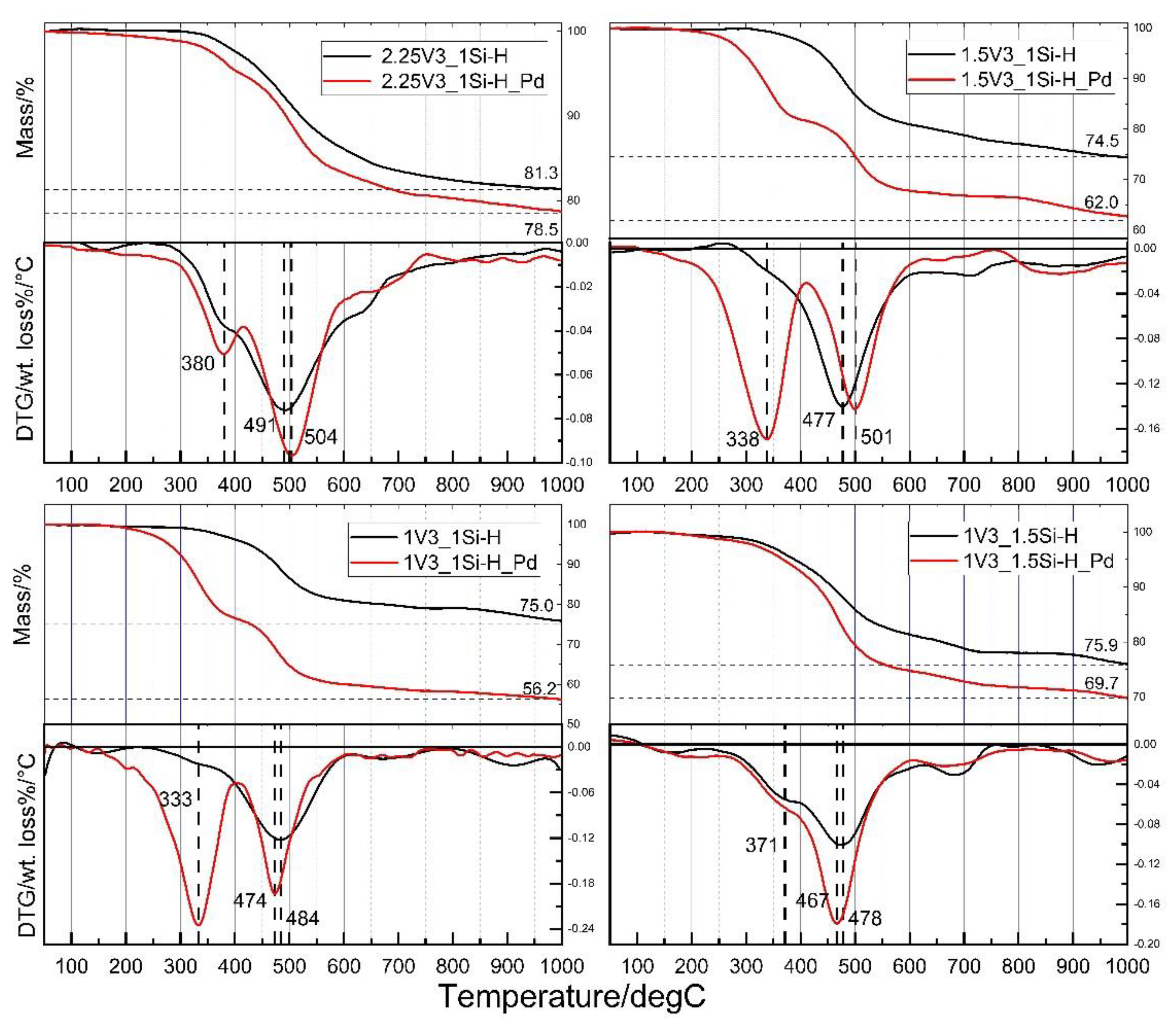
3.3. Thermal Investigations
4. Conclusions
- Pyrolysis of the V3 polymer-based polyHIPEs performed in Ar atmosphere at 1000 °C leads to Si-C-O materials composed of silicon oxycarbide and free carbon phases; the microstructure of the precursors is preserved in this process, ceramic yields are high, in the range of 74.5–81.3 mass %.
- V3 polymer-based polyHIPEs with incorporated metallic Pd nanoparticles, after pyrolysis carried out in Ar atmosphere at 1000 °C give Si-C-O ceramics, containing silicon oxycarbide and free carbon phases, with incorporated metallic Pd particles. Upon pyrolysis, sintering of the initial Pd nanoparticles occurs.
- Incorporation of Pd significantly affects thermal properties of the V3 polymer-based polyHIPEs. The materials with introduced Pd particles show lower thermal stability and give lower ceramic yields at 1000 °C (56.2–78.5 mass %) than the initial ones. Pd mainly influences the mechanism of bond redistributions at Si atoms that occur in the temperature range between 300 and 600 °C. The extent to which Pd modifies thermal properties of the V3 polymer-based polyHIPE is related to polymer cross-linking degree in the material.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mark, J.E. Some Interesting Things about Polysiloxanes. Acc. Chem. Res. 2004, 37, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Riedel, R.; Mera, G.; Hauser, R.; Kloczynski, A. Silicon-Based Polymer-Derived Ceramics: Synthesis Properties and Applications—A Review. J. Ceram. Soc. Jpn. 2006, 6, 425–444. [Google Scholar] [CrossRef] [Green Version]
- Arango-Ospina, M.; Xie, F.; Gonzalo-Juan, I.; Riedel, R.; Ionescu, E.; Boccaccini, A.R. Review: Silicon oxycarbide based materials for biomedical applications. Appl. Mater. Today 2020, 18, 100482. [Google Scholar] [CrossRef]
- Iastrenski, M.F.; da Silva, P.R.C.; Tarley, C.R.T.; Segatelli, M.G. Influence of molecular architecture of Si-containing precursors and HF chemical treatment on structural and textural features of silicon oxycarbide (SiOC) materials. Ceram. Int. 2019, 45, 21698–21708. [Google Scholar] [CrossRef]
- Soraru, G.D.; Dallapiccola, E.; D’Andrea, G. Mechanical Characterization of Sol–Gel-Derived Silicon Oxycarbide Glasses. J. Am. Ceram. Soc. 1996, 79, 2074–2080. [Google Scholar] [CrossRef]
- Walter, S.; Soraru, G.D.; Brequel, H.; Enzo, S. Microstructural and mechanical characterization of sol gel-derived Si–O–C glasses. J. Eur. Ceram. Soc. 2002, 22, 2389–2400. [Google Scholar] [CrossRef]
- Campostrini, R.; D’Andrea, G.; Carturan, G.; Ceccato, R.; Soraru, G.D. Pyrolysis study of methyl-substituted Si-H containing gels as precursors for oxycarbide glasses, by combined thermogravimetry, gas chromatographic and mass spectrometric analysis. J. Mater. Chem. 1996, 6, 585–594. [Google Scholar] [CrossRef]
- Camino, G.; Lomakinm, S.M.; Lageard, M. Thermal polydimethylsiloxane degradation. Part 2. The degradation mechanisms. Polymer 2002, 43, 2011–2015. [Google Scholar] [CrossRef]
- Fawcett, A.S.; Grande, J.B.; Brook, M.A. Rapid, metal-free room temperature vulcanization produces silicone elastomers. J. Polym. Sci. Part A Polym. Chem. 2012, 51, 644–652. [Google Scholar] [CrossRef]
- Baquey, G.; Moine, L.; Babot, O.; Degueil, M.; Maillard, B. Model study of the crosslinking of polydimethylsiloxanes by peroxides. Polymer 2005, 46, 6283–6292. [Google Scholar] [CrossRef]
- Deriabin, K.V.; Dobrynin, M.V.; Islamova, R.M. A metal-free radical technique for cross-linking of polymethylhydrosiloxane or polymethylvinylsiloxane using AIBN. Dalton Trans. 2020, 49, 8855–8858. [Google Scholar] [CrossRef] [PubMed]
- Nyczyk, A.; Paluszkiewicz, C.; Hasik, M.; Cypryk, M.; Pospiech, P. Cross-linking of linear vinylpolysiloxanes by hydrosilylation—FTIR spectroscopic studies. Vib. Spectrosc. 2012, 59, 1–8. [Google Scholar] [CrossRef]
- Colombo, P.; Mera, G.; Riedel, R.; Soraru, G.D. Polymer-Derived Ceramics: 40 Years of Research and Innovation in Advanced Ceramics. J. Am. Ceram. Soc. 2010, 93, 1805–1837. [Google Scholar] [CrossRef]
- Wójcik-Bania, M.; Olejarka, J.; Gumuła, T.; Łącz, A.; Hasik, M. Influence of metallic palladium on thermal properties of polysiloxane networks. Polym. Degrad. Stabil. 2014, 109, 249–260. [Google Scholar] [CrossRef]
- Wójcik-Bania, M.; Krowiak, A.; Strzezik, J.; Hasik, M. Pt supported on cross-linked poly(vinylsiloxanes) and SiCO ceramics—new materials for catalytic applications. Mater. Des. 2016, 96, 171–179. [Google Scholar] [CrossRef]
- Vakifahmetoglu, C.; Colombo, P.; Pauletti, A.; Martin, C.F.; Babonneau, F. SiOC Ceramic Monoliths with Hierarchical Porosity. Int. J. Appl. Ceram. Technol. 2010, 7, 528–535. [Google Scholar] [CrossRef]
- Biasetto, L.; Pena-Alonso, R.; Soraru, G.D.; Colombo, P. Etching of SiOC ceramic foams. Adv. Appl. Ceram. 2008, 107, 106–110. [Google Scholar] [CrossRef]
- Chauhan, P.K.; Sujith, R.; Parameshwaran, R.; Prasad, A.V.S.S. Role of polysiloxanes in the synthesis of aligned porous silicon oxycarbide ceramics. Ceram. Int. 2019, 45, 8150–8156. [Google Scholar] [CrossRef]
- Vakifahmetoglu, C.; Zeydanli, D.; Innocentini, M.; Ribeiro, F.d.S.; Lasso, P.R.O.; Soraru, G.D. Gradient-Hierarchic-Aligned Porosity SiOC Ceramics. Sci. Rep. 2017, 7, 41049. [Google Scholar] [CrossRef] [Green Version]
- Mrówka, J.; Gackowski, M.; Lityńska-Dobrzyńska, L.; Bernasik, A.; Kosydar, R.; Drelinkiewicz, A.; Hasik, M. Poly(methylvinylsiloxane)-Based High Internal Phase Emulsion-Templated Materials (polyHIPEs)—Preparation, Incorporation of Palladium, and Catalytic Properties. Ind. Eng. Chem. Res. 2020, 59, 19485–19499. [Google Scholar] [CrossRef]
- Kaur, S.; Mônego, G.; Rezwan, K.; Wilhelm, M. Synthesis of Porous Ni-SiC(O)-Based Nanoxomposites: Effect of Nickel Acetylacetonate and Poly(Ethylene Glycol) Methacrylate Modification on Specific Surface Area and Porosity. Adv. Eng. Mater. 2019, 22, 1901036. [Google Scholar] [CrossRef]
- Schumacher, D.; Wilhelm, M.; Rezwan, K. Porous SiOC monoliths with catalytic activity by in situ formation of Ni nanoparticles in solution-based freeze casting. J. Am. Ceram. Soc. 2020, 103, 2991–3001. [Google Scholar] [CrossRef]
- Adam, M.; Bäumer, M.; Schowalter, M.; Birkenstock, J.; Wilhelm, M.; Grathwohl, G. Generation of Pt- and Pt/Zn-containing ceramers and their structuring as macro/microporous foams. Chem. Eng. J. 2014, 247, 205–2015. [Google Scholar] [CrossRef]
- Canuto, A.S.T.; Mooste, M.; Kibena, P.E.; Matisen, L.; Merisalu, M.; Kook, M.; Sammelselg, V.; Tammeveski, K.; Wilhelm, M.; Rezwan, K. Polymer-Derived Co/Ni–SiOC(N) Ceramic Electrocatalysts for Oxygen Reduction Reaction in Fuel Cells. Catal. Sci. Technol. 2019, 9, 854–866. [Google Scholar] [CrossRef]
- Carnachan, R.J.; Bokhari, M.; Przyborski, S.A.; Cameron, N.R. Tailoring the morphology of emulsion-templated porous polymers. Soft Matter 2006, 2, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, M.S.; Cameron, N.R. (Eds.) PolyHIPEs—Porous Polymers from High Internal Phase Emulsions. In Encyclopedia of Polymer Science and Technology; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar] [CrossRef]
- Silverstein, M.S. PolyHIPEs: Recent advances in emulsion-templated porous polymers. Prog. Polym. Sci. 2013, 39, 199–234. [Google Scholar] [CrossRef]
- Zhang, T.; Sanguramath, R.A.; Israel, S.; Silverstein, M.S. Emulsion Templating: Porous Polymers and Beyond. Macromolecules 2019, 52, 5445–5479. [Google Scholar] [CrossRef] [Green Version]
- Grosse, M.-T.; Lamotte, M.; Birot, M.; Deleuze, H. Preparation of microcellular polysiloxane monoliths. J. Polym Sci. A Polym. Chem. 2007, 46, 21–32. [Google Scholar] [CrossRef]
- Cançado, L.G.; Takai, K.; Enoki, T. General equation for the determination of the crystallite size La of nanographite by Raman spectroscopy. Appl. Phys. Lett. 2006, 88, 163106. [Google Scholar] [CrossRef]
- Wen, Q.; Yu, Z.; Riedel, R. The fate and role of in situ formed carbon in polymer-derived ceramics. Prog. Mater. Sci. 2020, 109, 100623. [Google Scholar] [CrossRef]
- Souza, B.F.; Yoshida, I.V.P.; Ferrari, J.L.; Schiavon, M.A. Silicon oxycarbide glasses derived from polymeric networks with different molecular architecture prepared by hydrosilylation reaction. J. Mater. Sci. 2013, 48, 1911–1919. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 2000, 61, 20. [Google Scholar] [CrossRef] [Green Version]
- Mera, G.; Navrotsky, A.; Sen, S.; Kleebe, H.-J.; Riedel, R. Polymer-derived SiCN and SiOC ceramics—Structure and energetics at the nanoscale. J. Mater. Chem. A 2013, 1, 3826–3836. [Google Scholar] [CrossRef]
- Hourlier, D.; Venkatachalam, S.; Ammar, M.-R.; Blum, Y. Pyrolytic conversion of organopolysiloxanes. J. Anal. Appl. Pyrolysis 2017, 123, 296–306. [Google Scholar] [CrossRef]
- Venkatachalam, S.; Hourlier, D. Heat treatment of commercial Polydimethylsiloxane PDMS precursors: Part I. towards conversion of patternable soft gels into hard ceramics. Ceram. Int. 2019, 45, 6255–6262. [Google Scholar] [CrossRef]
- File 065-6174. The International Centre for Diffraction Data–ICDD. Available online: https://www.icdd.com (accessed on 27 July 2021).
- Hasik, M.; Wójcik-Bania, M.; Nyczyk, A.; Gumuła, T. Polysiloxane-POSS systems as precursors to SiCO ceramics. React. Funct. Polym. 2013, 73, 779–788. [Google Scholar] [CrossRef]
- Belot, V.; Corriu, R.J.P.; Leclercq, D.; Mutin, P.H.; Vioux, A. Thermal redistribution reactions in crosslinked polysiloxanes. J. Polym. Sci. A Polym. Chem. 1992, 30, 613–623. [Google Scholar] [CrossRef]
- Gualandris, V.; Bahloul-Hourlier, D.; Babonneau, F. Structural Investigation of the First Stages of Pyrolysis of Si-C-O Preceramic Polymers Containing Si–H Bonds. J. Sol-Gel Sci. Technol. 1999, 14, 39–48. [Google Scholar] [CrossRef]
- Bahloul-Hourlier, D.; Latournerie, J.; Dempsey, P. Reaction pathways during the thermal conversion of polysiloxane precursors into oxycarbide ceramics. J. Europ. Ceram. Soc. 2005, 25, 979–985. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
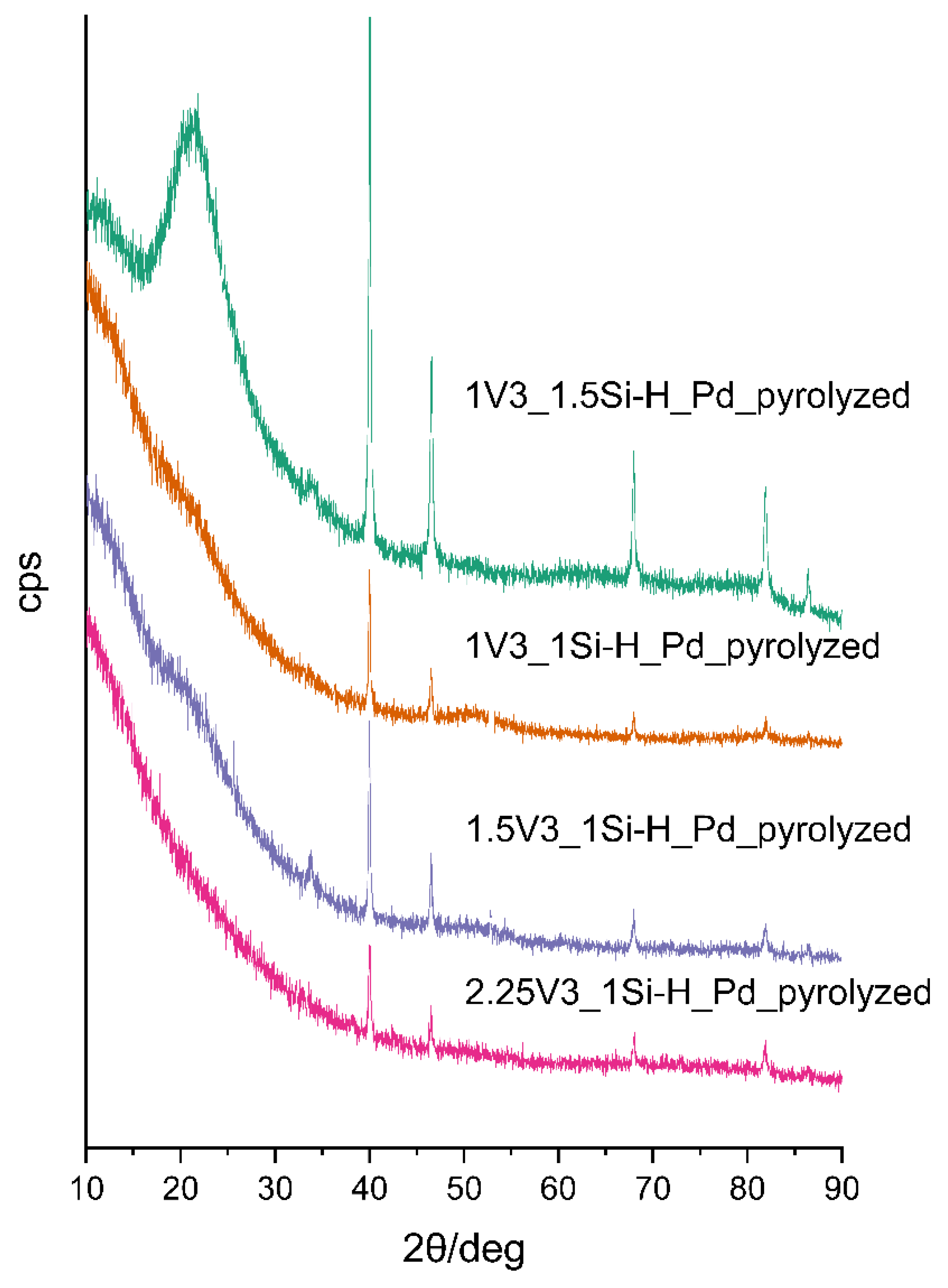{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Continuous Phase | Internal Phase | ||||
|---|---|---|---|---|---|---|
| V3 Polymer (g) | D4H (g) | DBE-224 (g) | Chlorobenzene (g) | Catalyst (μL) | 0.02M NaCl(aq) (g) | |
| 2.25V3_1Si-H | 1 | 0.31 | 0.33 | 0.41 | 7 | 9.32 |
| 1.5V3_1Si-H | 1 | 0.47 | 0.36 | 0.46 | 7 | 10.43 |
| 1V3_1Si-H | 1 | 0.70 | 0.43 | 0.53 | 7 | 12.10 |
| 1V3_1.5Si-H | 1 | 1.13 | 0.53 | 0.67 | 7 | 15.15 |
| Sample | Concentration/at % | C:O:Si Atomic Ratio (EDS) | ID/IG Ratio | Mean Carbon Domain Size in the Pyrolyzed Sample/nm | ||
|---|---|---|---|---|---|---|
| C | O | Si | ||||
| 2.25V3_1Si-H 2.25V3_1Si-Hpyrolyzed | 52.08 10.40 | 28.51 60.45 | 19.42 29.15 | 2.68:1.47:1 0.35:2.07:1 | − 2.48 | − 7 |
| 1.5V3_1Si-H 1.5V3_1Si-Hpyrolyzed | 50.54 6.84 | 27.14 60.80 | 22.33 32.36 | 2.26:1.22:1 0.21:1.88:1 | − 3.22 | − 6 |
| 1V3_1Si-H 1V3_1Si-Hpyrolyzed | 50.32 5.26 | 27.31 58.56 | 22.37 36.18 | 2.25:1.22:1 0.14:1.62:1 | − 2.48 | − 8 |
| 1V3_1.5Si-H 1V3_1.5Si-Hpyrolyzed | 47.58 8.05 | 27.07 57.97 | 25.35 33.98 | 1.88:1.07:1 0.24:1.71:1 | − 2.71 | − 7 |
| Sample | Pd Content/wt % 1 | Mean Pd Crystallite Size/nm 2 | ID/IG Ratio | Mean Carbon Domain Size/nm |
|---|---|---|---|---|
| 2.25V3_1Si-H_Pdpyrolyzed | 1.39 | 36.9 | 2.34 | 8 |
| 1.5V3_1Si-H_Pdpyrolyzed | 2.74 | 42.7 | 1.86 | 10 |
| 1V3_1Si-H_Pdpyrolyzed | 2.66 | 47.7 | 3.20 | 6 |
| 1V3_1.5Si-H_Pdpyrolyzed | 2.03 | 35.4 | 2.06 | 9 |
| Sample | T5/°C | T10 (T10−T5)/°C | Ceramic Yield at 1000 °C/mass% | Temperatures of the Fastest DecomPosition/°C |
|---|---|---|---|---|
| 2.25V3_1Si-H 2.25V3_1Si-H_Pd | 452 410 | 521 (69) 493 (83) | 81.3 78.5 | 380 (sh 1), 491 380, 504 |
| 1.5V3_1Si-H 1.5V3_1Si-H_Pd | 440 297 | 475 (35) 330 (33) | 74.5 62.0 | 477 338, 501 |
| 1V3_1Si-H 1V3_1Si-H_Pd | 426 280 | 477 (51) 313 (33) | 75.0 56.2 | 484 333, 474 |
| 1V3_1.5Si-H 1V3_1.5Si-H_Pd | 391 368 | 459 (68) 433 (65) | 75.9 69.7 | 371 (sh 1), 478 371 (sh 1), 467 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mrówka, J.; Partyka, J.; Hasik, M. Porous Materials Based on Poly(methylvinylsiloxane) Cross-Linked with 1,3,5,7-Tetramethylcyclotetrasiloxane in High Internal Phase Emulsion as Precursors to Si-C-O and Si-C-O/Pd Ceramics. Materials 2021, 14, 5661. https://doi.org/10.3390/ma14195661
Mrówka J, Partyka J, Hasik M. Porous Materials Based on Poly(methylvinylsiloxane) Cross-Linked with 1,3,5,7-Tetramethylcyclotetrasiloxane in High Internal Phase Emulsion as Precursors to Si-C-O and Si-C-O/Pd Ceramics. Materials. 2021; 14(19):5661. https://doi.org/10.3390/ma14195661
Chicago/Turabian StyleMrówka, Jan, Janusz Partyka, and Magdalena Hasik. 2021. "Porous Materials Based on Poly(methylvinylsiloxane) Cross-Linked with 1,3,5,7-Tetramethylcyclotetrasiloxane in High Internal Phase Emulsion as Precursors to Si-C-O and Si-C-O/Pd Ceramics" Materials 14, no. 19: 5661. https://doi.org/10.3390/ma14195661