
Effect of Microwave Treatment in a High Pressure Microwave Reactor on Graphene Oxide Reduction Process—TEM, XRD, Raman, IR and Surface Electron Spectroscopic Studies


 ,
, 
 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. TEM-PEELS and XRD Equipment
2.3. Raman Equipment
2.4. IR Equipment
2.5. XPS Equipment
3. Results and Discussion
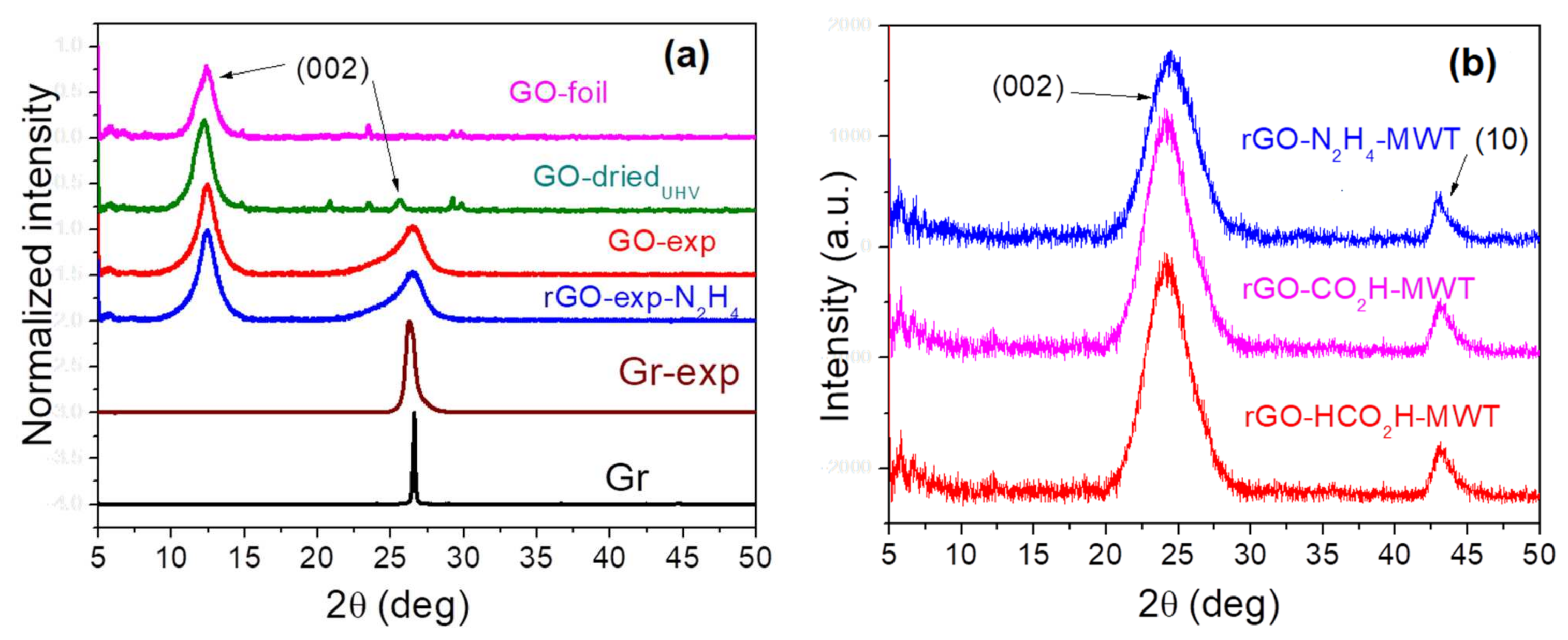
3.1. Structural Properties by TEM-PEELS and XRD
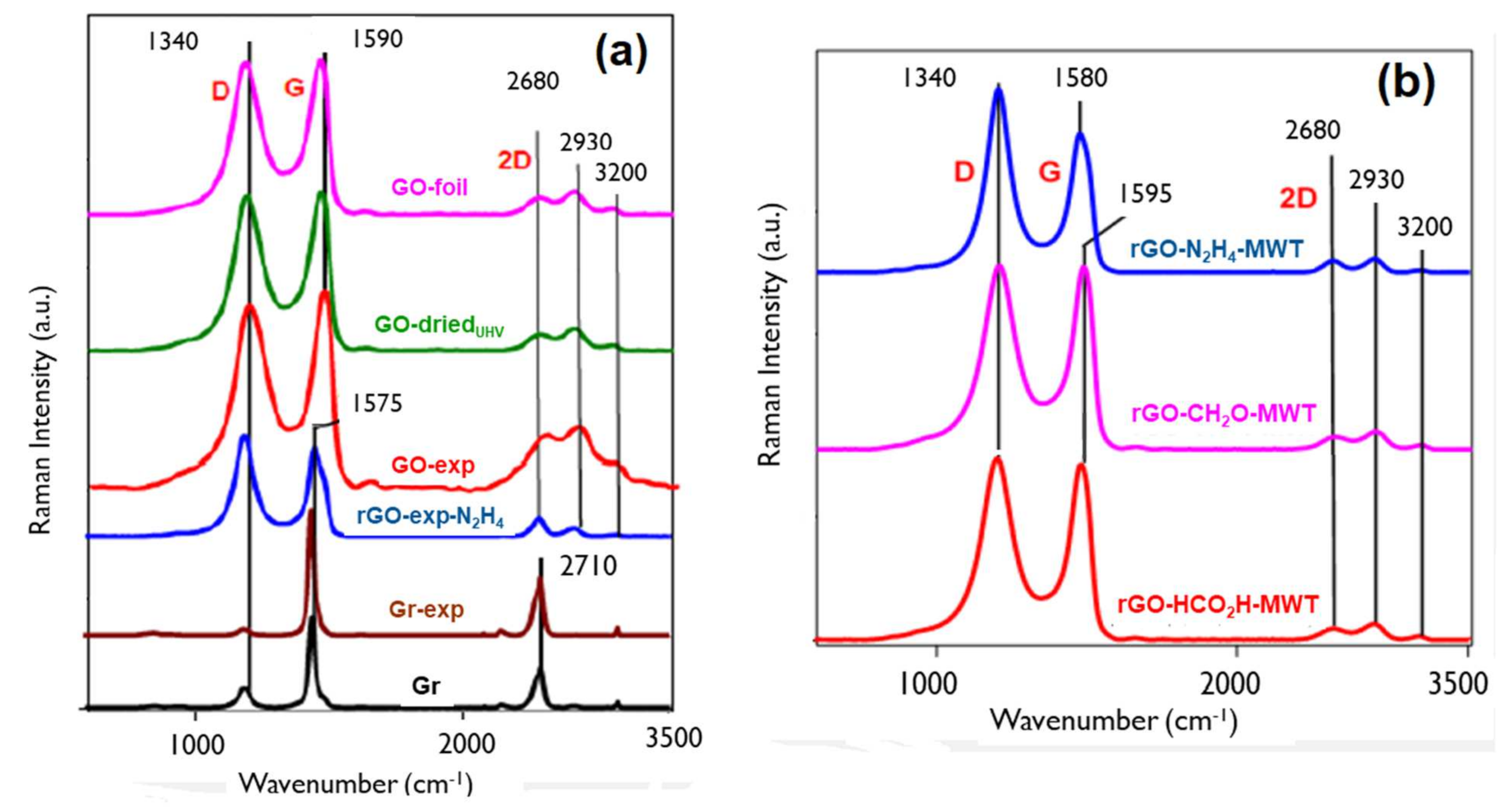
3.2. Bulk Physical and Structural Properties by Raman Spectroscopy
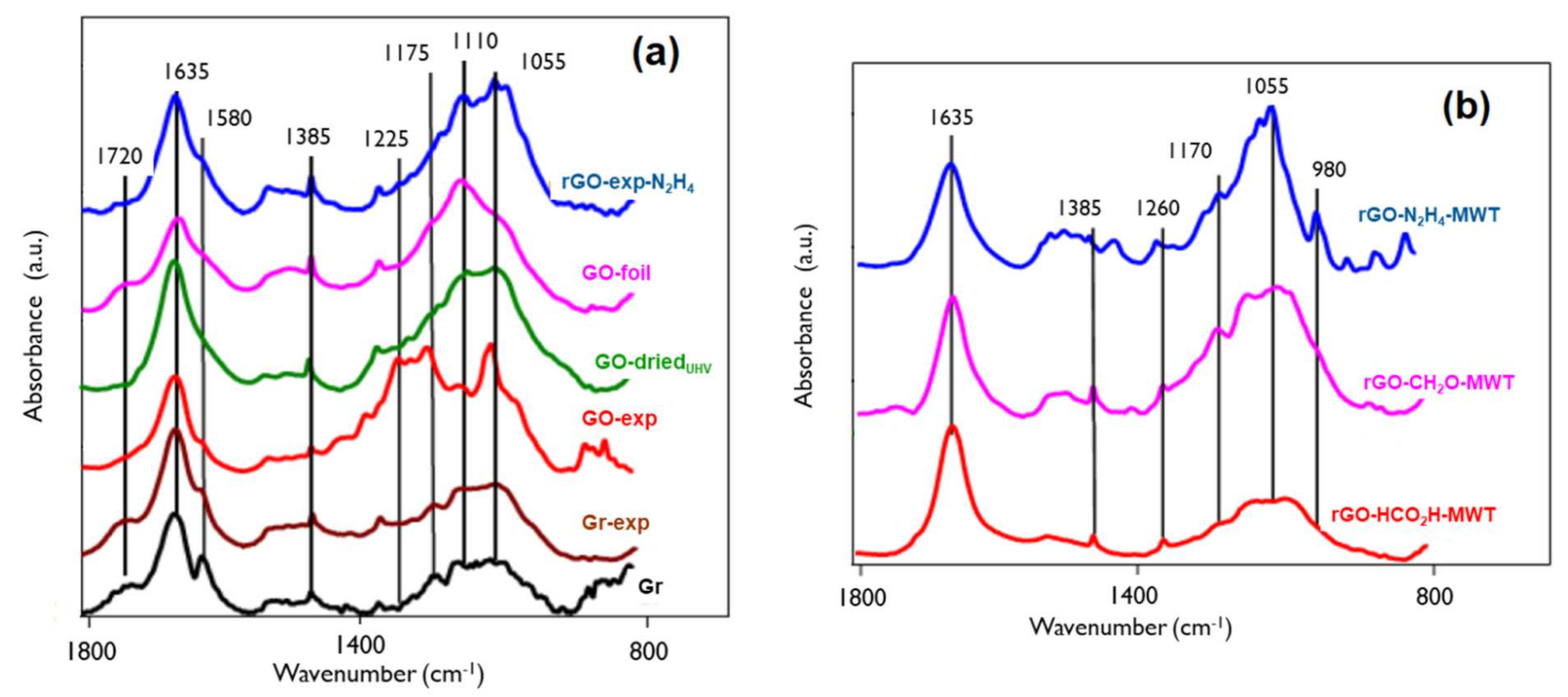
3.3. Bulk Chemical Properties by IR
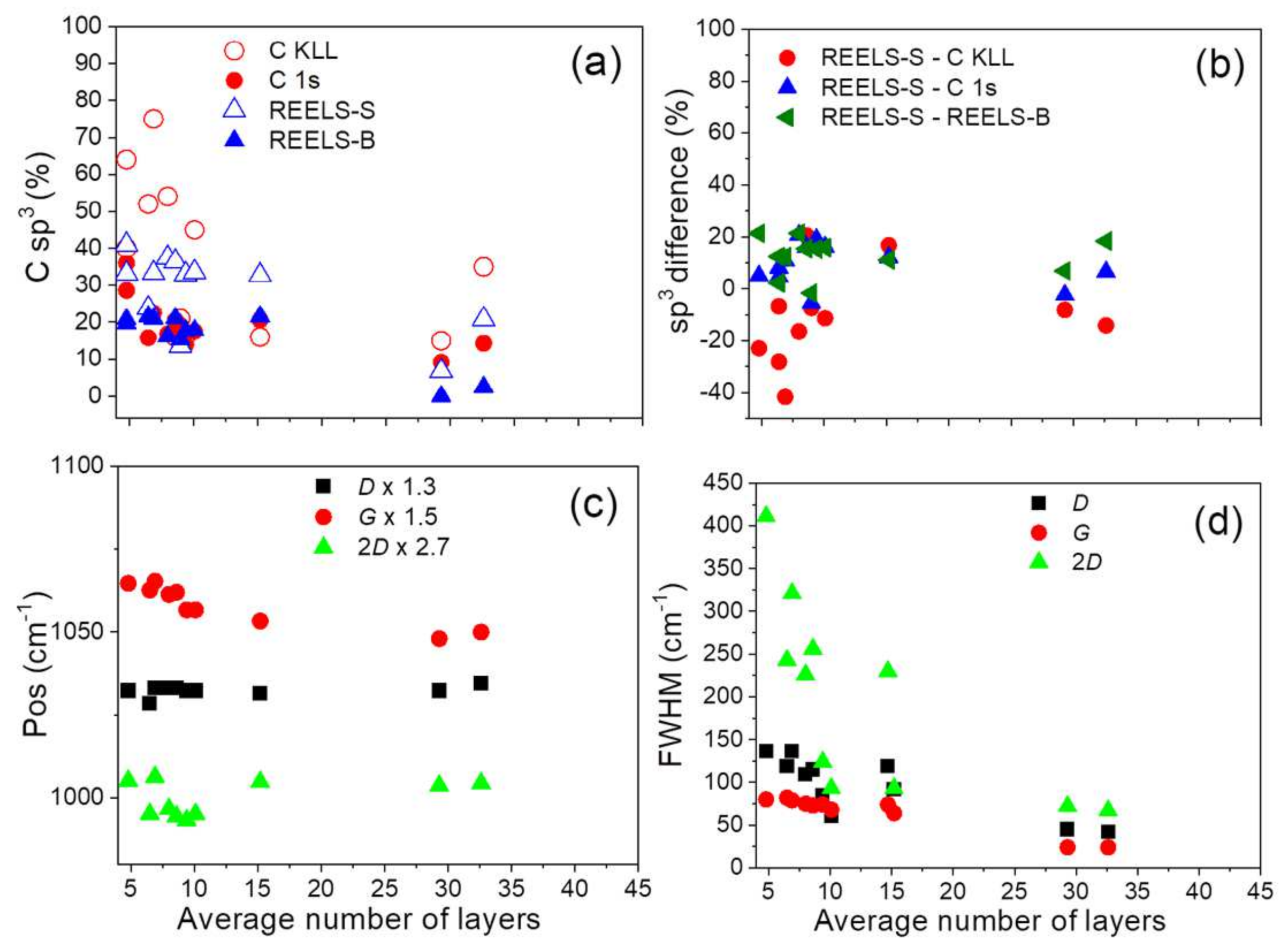
3.4. Evaluation of Surface Chemical and Structural Properties by REELS
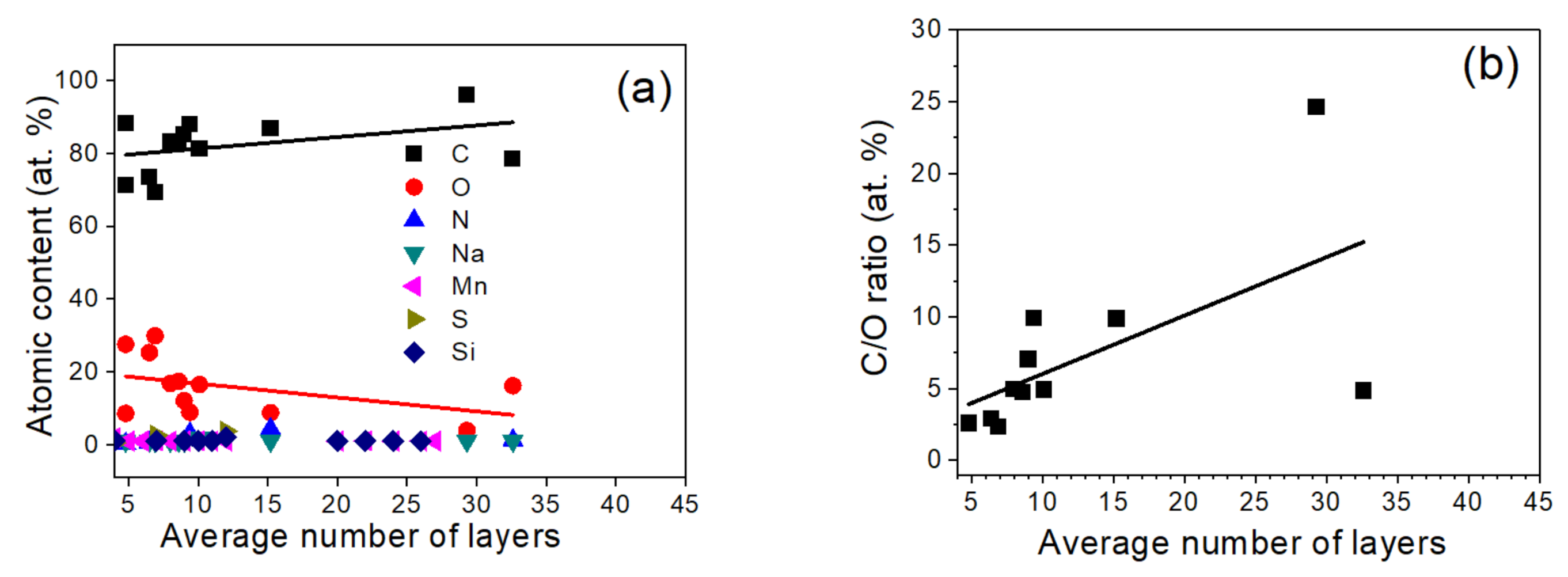
3.5. Surface Elemental Content by XPS
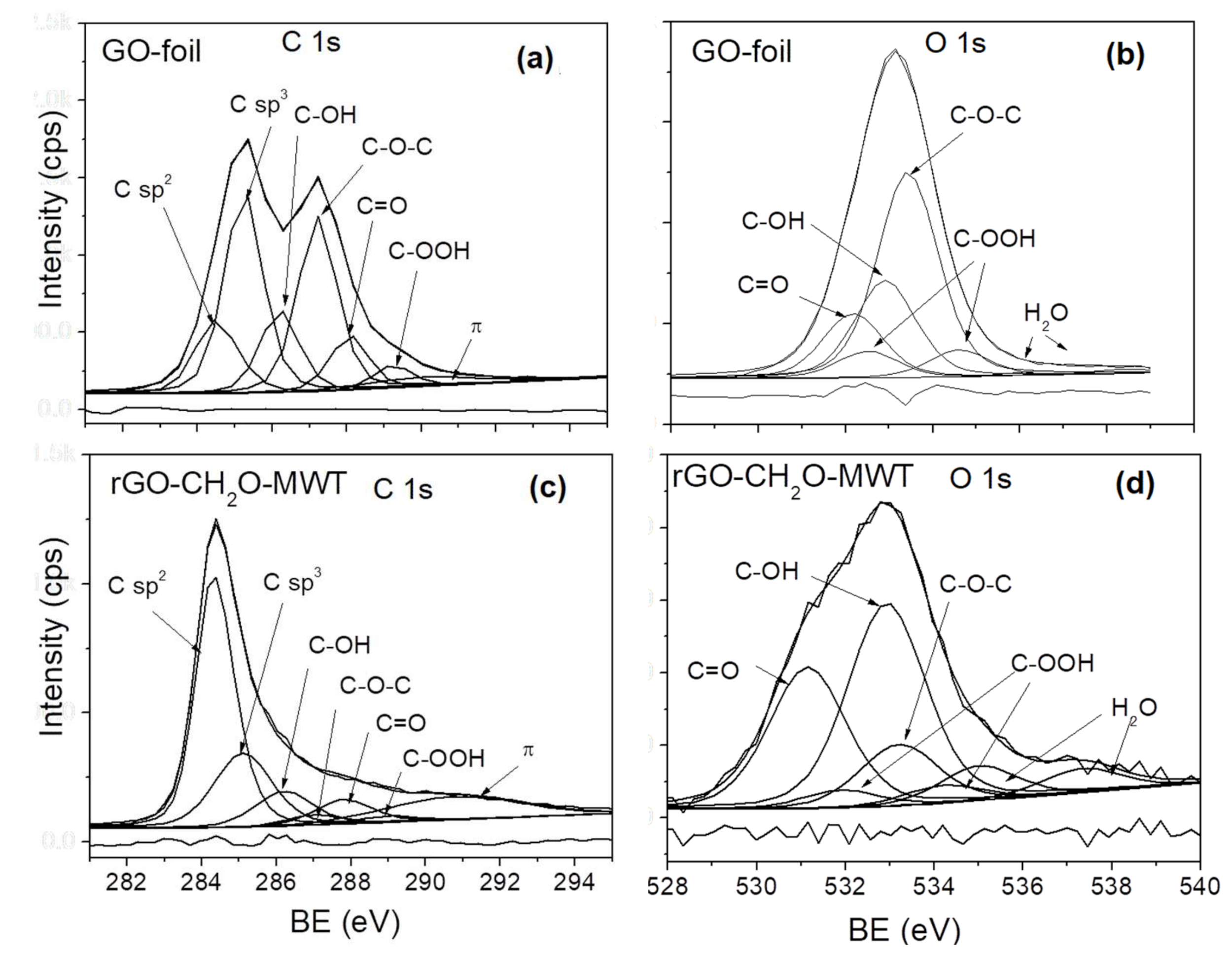
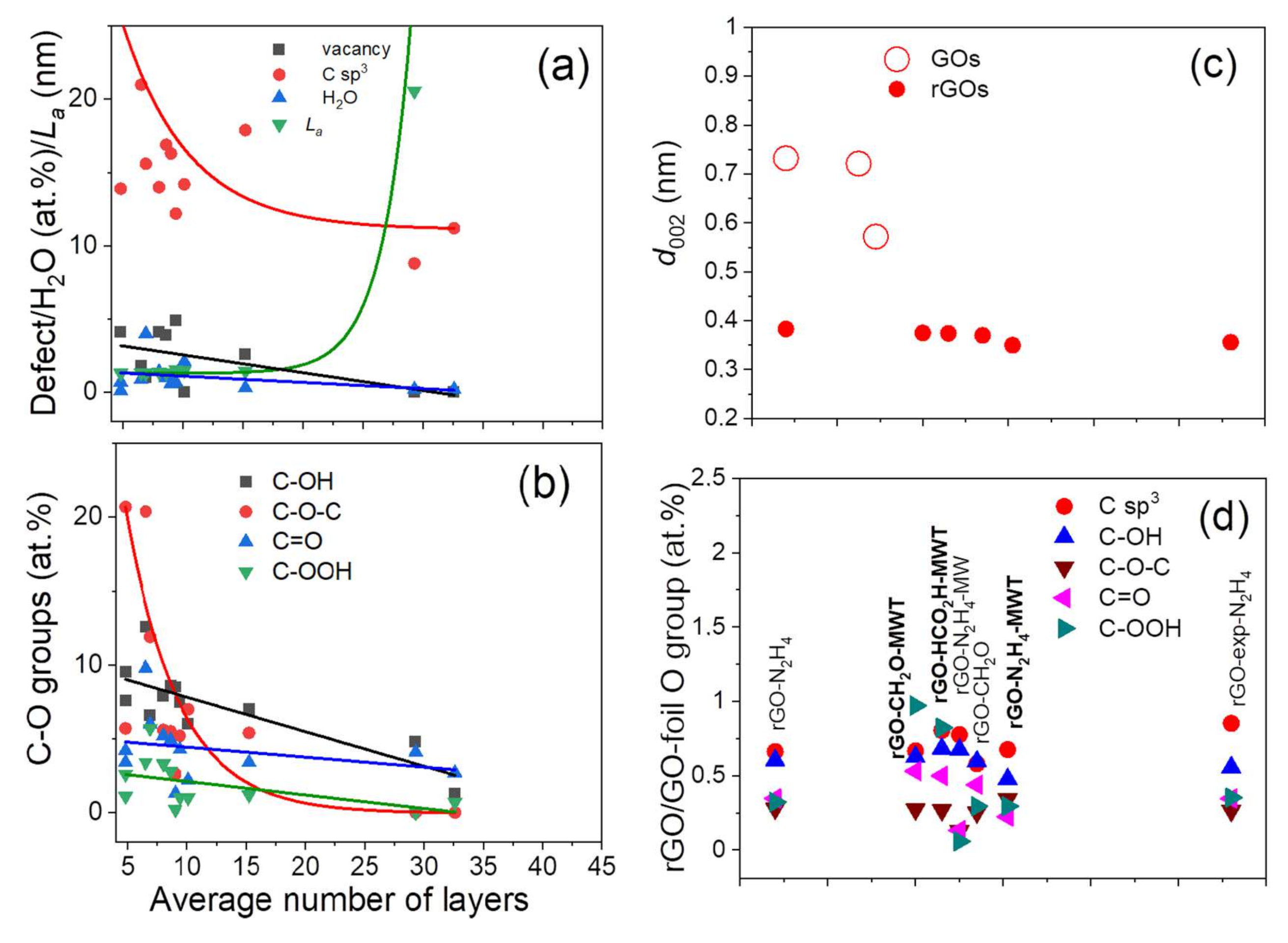
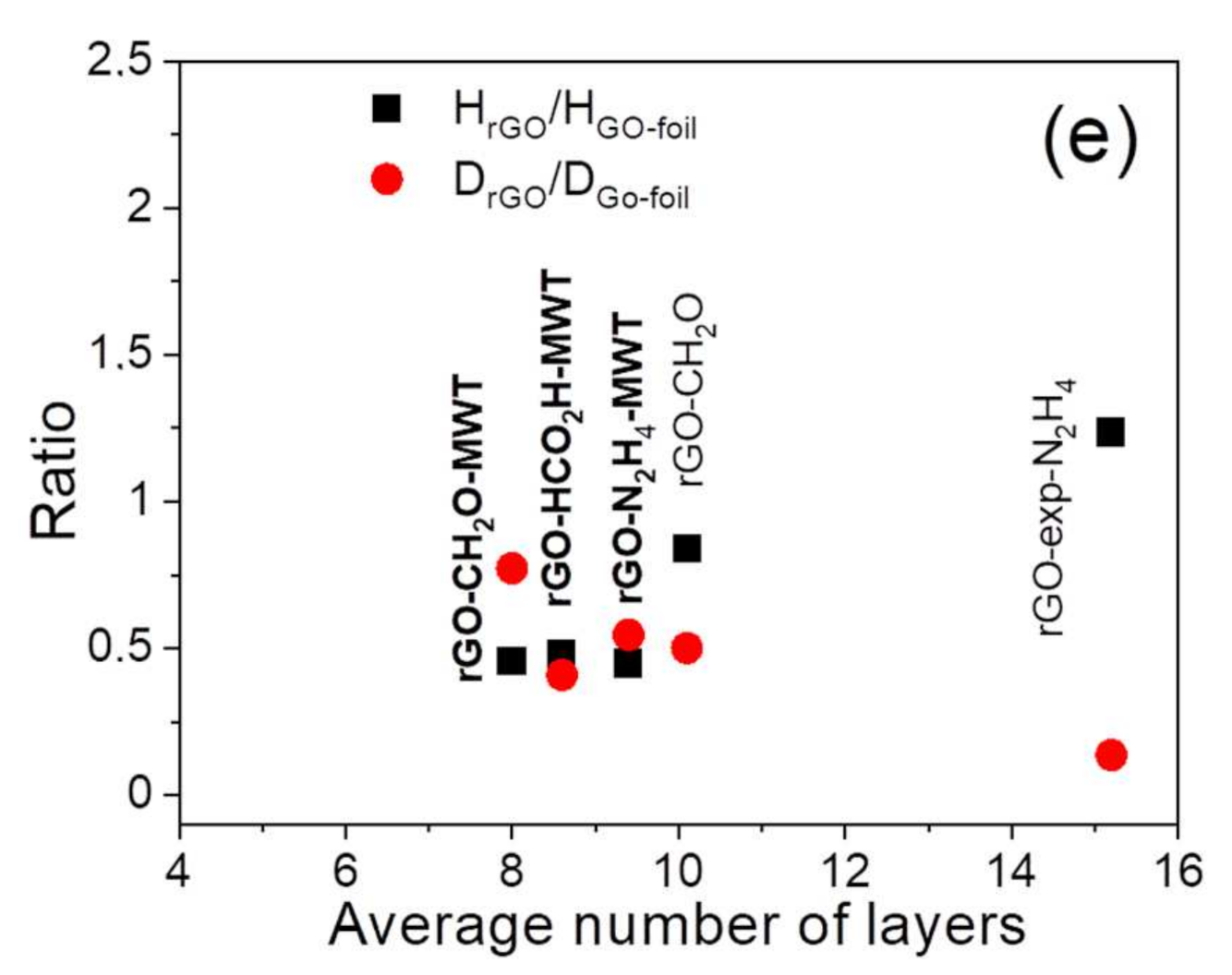
3.6. Surface Content of Carbon Hybridizations and Oxygen Groups by XPS and AES
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhuo, Q.; Mao, Y.; Lu, S.; Cui, B.; Yu, L.; Tang, J.; Sun, J.; Yan, C. Seed-assisted synthesis of graphene films on insulating substrate. Materials 2019, 12, 1376. [Google Scholar] [CrossRef] [Green Version]
- Kamedulski, P.; Ilnicka, A.; Lukaszewicz, J.P. Selected aspects of graphene exfoliation as an introductory step towards 3D structuring of graphene nano-sheets. Curr. Graphene Sci. 2018, 2, 106–117. [Google Scholar] [CrossRef]
- Prekodravac, J.R.; Kepić, D.P.; Colmenares, J.C.; Giannakoudakis, D.A.; Jovanović, S.P. A comprehensive review on selected graphene synthesis methods: From electrochemical exfoliation through rapid thermal annealing towards biomass pyrolysis. J. Mater. Chem. C 2021, 9, 6722–6748. [Google Scholar] [CrossRef]
- Jakhar, R.; Yap, J.E.; Joshi, R. Microwave reduction of graphene oxide. Carbon 2020, 170, 277–293. [Google Scholar] [CrossRef]
- Xie, X.; Zhou, Y.; Huang, K. Advances in microwave-assisted production of reduced graphene oxide. Front. Chem. 2019, 7, 335. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-R.; Lee, S.-H.; Lee, K.-H. Scalable production of large single-layers graphenes by microwave exfoliation in deionized water. Carbon 2018, 134, 431–438. [Google Scholar] [CrossRef]
- Wei, T.; Fan, Z.; Luo, G.; Zheng, C.; Xie, D. A rapid and efficient method to prepare exfoliated graphite by microwave irradiation. Carbon 2008, 50, 313–347. [Google Scholar] [CrossRef]
- Falcao, E.H.L.; Blair, R.G.; Mack, J.J.; Viculis, L.M.; Kwon, C.; Bendikov, M.; Kaner, R.B.; Dunn, B.S.; Wudl, F. Microwave exfoliation of a graphite intercalation compound. Carbon 2007, 45, 1364–1369. [Google Scholar] [CrossRef]
- Sridhar, V.; Jeon, J.-H.; Oh, I.-K. Synthesis of graphene nano-sheets using eco-friendly chemicals and microwave ratioation. Carbon 2010, 48, 2953–2957. [Google Scholar] [CrossRef]
- Tryba, B.; Morawski, A.W.; Inagaki, M. Preparation of exfoliated graphite by microwave irradiation. Carbon 2005, 45, 2397–2429. [Google Scholar] [CrossRef]
- Chen, W.; Yan, L.; Bangal, P.R. Preparation of graphene by the rapid and mild thermal reduction of graphene oxide induced by microwaves. Carbon 2010, 48, 1146–1152. [Google Scholar] [CrossRef]
- Zhu, Y.; Murali, S.; Stoller, M.D.; Velamakanni, A.; Piner, R.D.; Ruoff, R.S. Microwave assisted exfoliation and reduction of graphite oxide for ultracapacitors. Carbon 2010, 48, 2106–2122. [Google Scholar] [CrossRef]
- Yang, J.; Jo, M.R.; Kang, M.; Huh, Y.S.; Jung, H.; Kang, Y.-M. Rapid and controllable synthesis of nitrogen doped reduced graphene using microwave-assisted hydrothermal reaction for high power-density supercapacitors. Carbon 2014, 73, 106–113. [Google Scholar] [CrossRef]
- Zhao, Y.; He, J. Novel template-assisted microwave conversion of graphene oxide to graphene patterns: A reduction transfer mechanism. Carbon 2019, 148, 159–163. [Google Scholar] [CrossRef]
- Hu, H.; Zhao, Z.; Zhou, Q.; Gogotsi, Y.; Qiu, J. The role of microwave absorption on formation of graphene from graphite oxide. Carbon 2012, 50, 3267–3273. [Google Scholar] [CrossRef]
- Quan, B.; Liang, X.; Ji, G.; Lv, J.; Dai, S.S.; Xu, G.; Du, Y. Laminated graphene oxide-supported high-efficiency microwave absorber fabricated by an in situ growth approach. Carbon 2018, 129, 310–320. [Google Scholar] [CrossRef]
- Canal-Rodríques, M.; Arenillas, A.; Menéndez, J.A.; Beneroso, D.; Rey-Raap, N. Carbon xerogels graphitised by microwave heating as anode materials in lithium-ion batteries. Carbon 2018, 137, 384–394. [Google Scholar] [CrossRef]
- Sridhar, V.; Jeon, J.-H.; Oh, I.-K. Microwave extraction of graphene from carbon fibres. Carbon 2011, 49, 222–226. [Google Scholar] [CrossRef]
- Kumar, R.; Savu, R.; Singh, R.K.; Joanni, E.; Singh, D.P.; Tiwari, V.S.; Vaz, A.R.; da Silva, E.T.S.G.; Maluta, J.R.; Kubota, L.T.; et al. Controlled density of defects assisted perforated structure in reduced graphene oxide nanosheets-palladium hybrids for enhanced ethanol electro-oxidation. Carbon 2017, 117, 137–146. [Google Scholar] [CrossRef]
- Kuang, B.; Song, W.; Ning, M.; Li, J.; Zhao, Z.; Guo, D.; Cao, M.; Jin, H. Chemical reduction dependent dielectric properties and dielectric loss mechanism of reduced graphene oxide. Carbon 2018, 127, 209–217. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 311–336. [Google Scholar] [CrossRef]
- Stobinski, L.; Lesiak, B.; Malolepszy, A.; Mazurkiewicz, M.; Mierzwa, B.; Zemek, J.; Jiricek, P.; Bieloshapka, I. Graphene oxide and reduced graphene oxide studied by the XRD, TEM and electron spectroscopy methods. J. Electron Spectrosc. Rel. Phenom. 2014, 195, 145–154. [Google Scholar] [CrossRef]
- Lesiak, B.; Trykowski, G.; Tóth, J.; Biniak, S.; Kövér, L.; Rangam, N.; Stobinski, L.; Malolepszy, A. Chemical and structural properties of reduced graphene oxide–dependence on the reducing agent. J. Mat. Sci. 2021, 56, 3738–3754. [Google Scholar] [CrossRef]
- Kövér, L.; Varga, D.; Cserny, I.; Tóth, J.; Tőkési, J. Some applications of high-energy, high-resolution Auger-electron spectroscopy using Bremsstrahlung radiation. Surf. Interface Anal. 1992, 19, 9–15. [Google Scholar] [CrossRef]
- Warren, B.E. X-Ray Di8raction in Random Layer Lattices. Phys. Rev. 1941, 59, 693–698. [Google Scholar] [CrossRef]
- Malard, L.M.; Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S. Raman spectroscopy in graphene. Phys. Rep. 2019, 473, 51–87. [Google Scholar] [CrossRef]
- Qi, Z.; Zhu, X.; Jin, H.; Zhang, T.; Kong, X.; Ruoff, R.S.; Qiao, Z.; Ji, H. Rapid identification of the layer number of large-area-graphene on copper. Chem. Matter. 2018, 30, 2067–2073. [Google Scholar] [CrossRef]
- Szirmai, P.; Márkus, B.G.; Chacón-Torres, J.C.; Eckerlein, P.; Edelthalhammer, K.; Englert, J.M.; Mundloch, U.; Hirsch, A.; Hauke, F.; Náfrádi, B.; et al. Characterizing the maximum number of layers in chemically exfoliated graphene. Sci. Rep. 2019, 9, 19480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, A.C.; Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 2000, 61, 14095–14107. [Google Scholar] [CrossRef] [Green Version]
- Tunistra, F.; Koenig, J.L. Raman spectrum of graphite. J. Chem. Phys. 1970, 53, 1126–1130. [Google Scholar] [CrossRef] [Green Version]
- Casiraghi, C.; Pisana, S.; Novoselov, K.S.; Geim, A.K.; Ferrari, A.C. Raman fingerprint of charged impurities in graphene. Appl. Phys. Lett. 2007, 91, 233108. [Google Scholar] [CrossRef] [Green Version]
- Eckmann, A.; Felten, A.; Mishchenko, A.; Britnell, L.; Krupke, R.; Novoselov, K.S.; Casiraghi, C. Probing nature of defects in graphene by Raman spectroscopy. Nano Lett. 2012, 12, 3925–3930. [Google Scholar] [CrossRef] [Green Version]
- Lucchese, M.M.; Stavale, F.; Martin Ferreire, E.H.; Vilani, C.; Moutinho, M.V.O.; Capaz, R.B.; Achete, C.A.; Jorio, A. Quantifying ion-induced defects and Raman relaxation length in graphene. Carbon 2010, 48, 1592–1597. [Google Scholar] [CrossRef]
- Cançado, L.G.; Jorio, A.; Martins Ferreira, E.H.; Sravale, F.; Achete, C.A.; Capaz, R.B.; Moutinho, M.V.O.; Lombardo, A.; Kulmala, T.S.; Ferrari, A.C. Quantifying defects in graphene via Raman Spectroscopy at different excitation energies. Nano Lett. 2011, 11, 3190–3196. [Google Scholar] [CrossRef] [Green Version]
- Ren, P.-G.; Yan, D.-X.; Ji, X.; Che, T.; Li, Z.-M. Temperature dependence of graphene oxide reduced by hydrazine hydrate. Nanotechnology 2011, 22, 055705. [Google Scholar] [CrossRef]
- Guo, H.; Peng, M.; Zhu, Z.; Sun, L. Preparation of reduced graphene oxide by infrared irradiation induced photothermal reduction. Nanoscale 2013, 5, 9040–9048. [Google Scholar] [CrossRef]
- Robertson, J. Diamond-like amorphous carbon. Mat. Sci. Eng. R 2002, 37, 129–281. [Google Scholar] [CrossRef] [Green Version]
- Calliari, L.; Fanczenko, S.; Filippi, M. Plasmon features in electron energy loss spectra from carbon materials. Carbon 2007, 45, 1410–1418. [Google Scholar] [CrossRef]
- Eberlein, J.T.; Bangert, U.; Nair, R.R.; Jones, R.; Gass, M.; Bleloch, A.L.; Novoselov, K.S.; Geim, A.; Briddon, P.R. Plasmon spectroscopy of free-standing graphene films. Phys. Rev. B Condens. Matter. 2008, 77, 233406–233414. [Google Scholar] [CrossRef] [Green Version]
- Johari, P.; Shenoy, V.B. Modulating optical properties of graphene oxide: Role of prominent functional groups. ACS Nano 2011, 5, 7640–7647. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, A.; Powell, C.J. Information depth for elastic-peak electron spectroscopy. Surf. Sci. 2004, 551, 106–124. [Google Scholar] [CrossRef]
- Jablonski, A.; Powell, C.J. Practical expressions for the mean escape depth, the information depth, and the effective attenuation length in Auger-electron spectroscopy and x-ray photoelectron spectroscopy. J. Vac. Sci. Technol. A 2009, 27, 253–261. [Google Scholar] [CrossRef]
- Jablonski, A.; Zemek, J. Overlayer thickness determination by XPS using the multiline approach. Surf. Interface Anal. 2009, 41, 193–204. [Google Scholar] [CrossRef]
- Touggard, S. Background Analysis of XPS/AES QUASES Simple BackgroundsVer. 2.2, 1999–2001 Tougaard Inc. Available online: http://www.quases.com (accessed on 31 August 2021).
- Mohai, M. XPS MultiQuant: Multimodel XPS quantification software. Surf. Interface Anal. 2004, 36, 828–832. [Google Scholar] [CrossRef]
- Mohai, M. Multimodel of X-ray photoelectron spectroscopy qunatification program for 32-bit Windows, XPSMuliQuant, ver. 7. 1999–2001.
- Scofield, H. Hartree-Slater Subshell Photoionization Cross-sections at 1254 and 1487 eV. J. Electron Spectrosc. Relat. Phenom. 1976, 8, 129–137. [Google Scholar] [CrossRef]
- Kwok, R.W.M. XPS Peak Fitting Program for WIN95/98 XPSPEAK, ver. 4.1, Department of Chemistry, The Chinese University of Hong Kong, [email protected]. Available online: http://www.uksaf.org/software.html>XPSPEAK4.1 (accessed on 31 August 2021).
- Lesiak, B.; Kövér, L.; Tóth, J.; Zemek, J.; Jiricek, P.; Kromka, A.; Rangam, N. C sp2/sp3 hybridisations in carbon nanomaterials–XPS and (X) AES study. Appl. Surf. Sci. 2018, 452, 223–231. [Google Scholar] [CrossRef]
- Butenko, Y.V.; Krishnamurthy, S.; Chakraborty, A.K.; Kuznetsov, V.L.; Dhanak, V.R.; Hunt, M.C.; Šiller, L. Photoemission study of onionlike carbons produced by annealing nanodiamonds. Phys. Rev. B 2005, 71, 075420. [Google Scholar] [CrossRef] [Green Version]
- Shim, S.H.; Kim, K.T.; Lee, J.U.; Jo, W.H. Facile method to functionalize graphene oxide and its application to poly (ethylene terephthalate)/graphene composite. ACS Appl. Mater. Interfaces 2012, 4, 4184–4191. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Yamada, Y.; Koinuma, M.; Sata, S. Origins of sp3C peaks in C1s X-ray photoelectron spectra of carbon materials. Anal. Chem. 2016, 88, 6110–6114. [Google Scholar] [CrossRef] [Green Version]
- Winter, B.; Aziz, E.F.; Hergenhahn, U.; Faubel, M.; Hertel, I.V. Hydrogen bonds in liquid water studied by electron spectroscopy. J. Chem. Phys. 2007, 126, 124504. [Google Scholar] [CrossRef]
- Yamamoto, S.; Bluhm, H.; Andersson, K.; Ketteler, G.; Ogasawara, H.; Salmeron, M.; Nilsson, A. In situ x-ray photoelectron spectroscopy studies of water on metals and oxides at ambient conditions. J. Phys. Condend. Matter. 2008, 20, 184025. [Google Scholar] [CrossRef] [Green Version]
- Estrade-Szwarckopf, H. XPS photoemission in carbonaceous materials: A ‘‘defect’’ peak beside the graphitic asymmetric peak. Carbon 2004, 42, 1713–1721. [Google Scholar] [CrossRef]
- Stobinski, L.; Lesiak, B.; Zemek, J.; Jiricek, P.; Biniak, S.; Trykowski, G. Studies of oxidized carbon nanotubes in temperature range RT-630oC by the infrared and electron spectroscopies. J. Alloy. Compd. 2010, 505, 379–384. [Google Scholar] [CrossRef]
- Lesiak, B.; Zemek, J.; Jiricek, P.; Stobinski, L. Temperature modification of oxidized multiwall carbon nantubes studied by electron spectroscopy methods. Phys. Status Solidi 2009, 246, 2645–2649. [Google Scholar] [CrossRef]
- Shinotsuka, H.; Tanuma, S.; Powell, C.J.; Penn, D.R. Calculations of electron inelastic mean free pahs. X. Data for 41 elemental solids over the 50 eV to 200 keV range with the relativistic full Penn algorithm. Surf. Interface Anal. 2015, 47, 871–888. [Google Scholar] [CrossRef]
- Lascovich, C.; Scaglione, S. Comparison among XAES, PEELS and XPS techniques for evaluation of Sp2 percentage in a-C:H. Appl. Surf. Sci. 1994, 78, 17–23. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Peak (002) | Peak (10) | ||
|---|---|---|---|---|
| Average d (nm) | Average H (nm) | Average n | Average D (nm) | |
| * GO-foil | 0.721 | 4.883 | 6.8 | * 22 |
| GO-driedUHV | 0.732 | 4.625 | 6.3 | - |
| * rGO-N2H4 | 0.383 | 1.302 | 3.4 | 8 |
| rGO-N2H4-MWT | 0.370 | 2.188 | 5.9 | 12 |
| GO-exp | 0.572 | 4.288 | 7.5 | - |
| rGO-exp-N2H4 | 0.356 | 6.028 | 16.9 | 3 |
| * rGO-CH2O | 0.350 | 4.099 | 11.7 | 11 |
| rGO-CH2O-MWT | 0.375 | 2.217 | 5.9 | 17 |
| rGO-HCO2H-MWT | 0.374 | 2.349 | 6.3 | 9 |
| * Gr | 0.344 | - | - | - |
| * Gr-exp | 0.347 | - | - | - |
| Sample | D Band | G Band | 2D Band | I(D)/ I(G) | I(2D)/ I(G) | La (nm) | |||
|---|---|---|---|---|---|---|---|---|---|
| Pos (cm−1) | FWHM (cm−1) | Pos (cm−1) | FWHM (cm−1) | Pos (cm−1) | FWHM (cm−1) | ||||
| * GO-foil | 1337 | 119 | 1594 | 82 | 2687 | 243 | 0.991 | 0.465 | 1.34 |
| GO-driedUHV | 1342 | 136 | 1597 | 80 | 2714 | 411 | 0.927 | 0.2611 | 1.30 |
| rGO-N2H4-MWT | 1342 | 85 | 1585 | 74 | 2682 | 124 | 1.32 | 0.0833 | 1.55 |
| GO-exp | 1343 | 136 | 1598 | 79 | 2717 | 321 | 0.928 | 0.2632 | 1.30 |
| rGO-exp-N2H4 | 1341 | 92 | 1580 | 64 | 2713 | 93 | 1.139 | 0.2033 | 1.44 |
| * rGO-CH2O | 1342 | 60 | 1585 | 68 | 2687 | 93 | 1.242 | 0.7210 | 1.5 |
| rGO-CH2O-MWT | 1343 | 109 | 1592 | 75 | 2691 | 226 | 1.036 | 0.0635 | 1.37 |
| rGO-HCO2H-MWT | 1343 | 115 | 1593 | 73 | 2685 | 256 | 1.002 | 0.5884 | 1.35 |
| * Gr | 1342 | 45 | 1572 | 24 | 2710 | 72 | 0.214 | 0.4315 | 20.56 |
| * Gr-exp | 1345 | 42 | 1575 | 24 | 2712 | 67 | 0.041 | 0.4604 | 107.32 |
| Sample | π + σ Contributions (%) | Average Number of Layers, n | |||||
|---|---|---|---|---|---|---|---|
| C sp2S Eloss = 19.5 eV | C sp2B Eloss = 27 eV | C sp3S Eloss = 23 eV | Csp3B Eloss = 33.8 eV | Csp2B/Csp2S | d from XRD (Table 1) | Average Value of n (XRD and REELS) | |
| * GO-foil | 13.0 | 33.0 | 33.2 | 20.8 | 2.54 | 6.1 | 6.5 ± 0.4 |
| GO-driedUHV | 15.9 | 23.4 | 41.0 | 19.7 | 1.47 | 3.2 | 4.8 ± 2.2 |
| ** rGO-N2H4 | 15.6 | 39.0 | 23.8 | 21.6 | 2.51 | 6.1 | 4.8 ± 1.9 |
| ** rGO-N2H4 -MW | 10.6 | 34.5 | 13.6 | 15.3 | 3.26 | 9.0 | 9.0 |
| rGO-N2H4-MWT | 13.4 | 35.9 | 33.0 | 17.7 | 2.67 | 12.8 | 9.4 ± 4.9 |
| GO-exp | 14.5 | 31.2 | 33.3 | 21.0 | 2.15 | 6.2 | 6.9 ± 0.9 |
| rGO-exp-N2H4 | 12.4 | 33.3 | 32.7 | 21.6 | 2.69 | 13.4 | 15.2 ± 2.5 |
| * rGO-CH2O | 17.2 | 31.4 | 33.6 | 17.8 | 1.83 | 8.4 | 10.1 ± 2.3 |
| rGO-CH2O-MWT | 14.3 | 32.0 | 37.5 | 16.2 | 2.24 | 10.0 | 8.0 ± 2.9 |
| rGO-HCO2H-MWT | 12.5 | 29.8 | 36.5 | 21.1 | 2.39 | 10.9 | 8.6 ± 3.3 |
| * Gr | 18.5 | 74.7 | 6.8 | 0 | 4.04 | 29.3 | |
| * Gr-exp | 14.7 | 61.9 | 20.8 | 2.5 | 4.21 | 32.6 | |
| Sample | Parameter D (eV) | C sp3 (%) (C KLL) | C sp3 (%) (C 1s) |
|---|---|---|---|
| * GO-foil | 19.1 | 40 | 28.6 |
| GO-driedUHV | 16.8 | 64 | 36 |
| ** rGO-N2H4 | 18.0 | 52 | 15.8 |
| ** rGO-N2H4-MW | 21.0 | 21 | 19.1 |
| rGO-N2H4-MWT | 21.5 | 16 | 13.9 |
| GO-exp | 15.7 | 75 | 22.5 |
| rGO-exp-N2H4 | 21.5 | 16 | 20.6 |
| * rGO-CH2O | 18.7 | 45 | 17.5 |
| rGO-CH2O-MWT | 17.8 | 54 | 16.8 |
| rGO-HCO2H-MWT | 21.5 | 16 | 20.5 |
| * Gr | 21.6 | 15 | 9.2 |
| * Gr-exp | 19.6 | 35 | 14.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lesiak, B.; Trykowski, G.; Tóth, J.; Biniak, S.; Kövér, L.; Rangam, N.; Małolepszy, A.; Stobiński, L. Effect of Microwave Treatment in a High Pressure Microwave Reactor on Graphene Oxide Reduction Process—TEM, XRD, Raman, IR and Surface Electron Spectroscopic Studies. Materials 2021, 14, 5728. https://doi.org/10.3390/ma14195728
Lesiak B, Trykowski G, Tóth J, Biniak S, Kövér L, Rangam N, Małolepszy A, Stobiński L. Effect of Microwave Treatment in a High Pressure Microwave Reactor on Graphene Oxide Reduction Process—TEM, XRD, Raman, IR and Surface Electron Spectroscopic Studies. Materials. 2021; 14(19):5728. https://doi.org/10.3390/ma14195728
Chicago/Turabian StyleLesiak, Beata, Grzegorz Trykowski, József Tóth, Stanisław Biniak, László Kövér, Neha Rangam, Artur Małolepszy, and Leszek Stobiński. 2021. "Effect of Microwave Treatment in a High Pressure Microwave Reactor on Graphene Oxide Reduction Process—TEM, XRD, Raman, IR and Surface Electron Spectroscopic Studies" Materials 14, no. 19: 5728. https://doi.org/10.3390/ma14195728
APA StyleLesiak, B., Trykowski, G., Tóth, J., Biniak, S., Kövér, L., Rangam, N., Małolepszy, A., & Stobiński, L. (2021). Effect of Microwave Treatment in a High Pressure Microwave Reactor on Graphene Oxide Reduction Process—TEM, XRD, Raman, IR and Surface Electron Spectroscopic Studies. Materials, 14(19), 5728. https://doi.org/10.3390/ma14195728