
Effect of Ruthenium and Cerium Oxide (IV) Promotors on the Removal of Carbon Deposit Formed during the Mixed Methane Reforming Process

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Catalyst Preparation
2.2. Methods and Instruments
3. Results
3.1. The Specific Surface Area of the Catalytic Materials
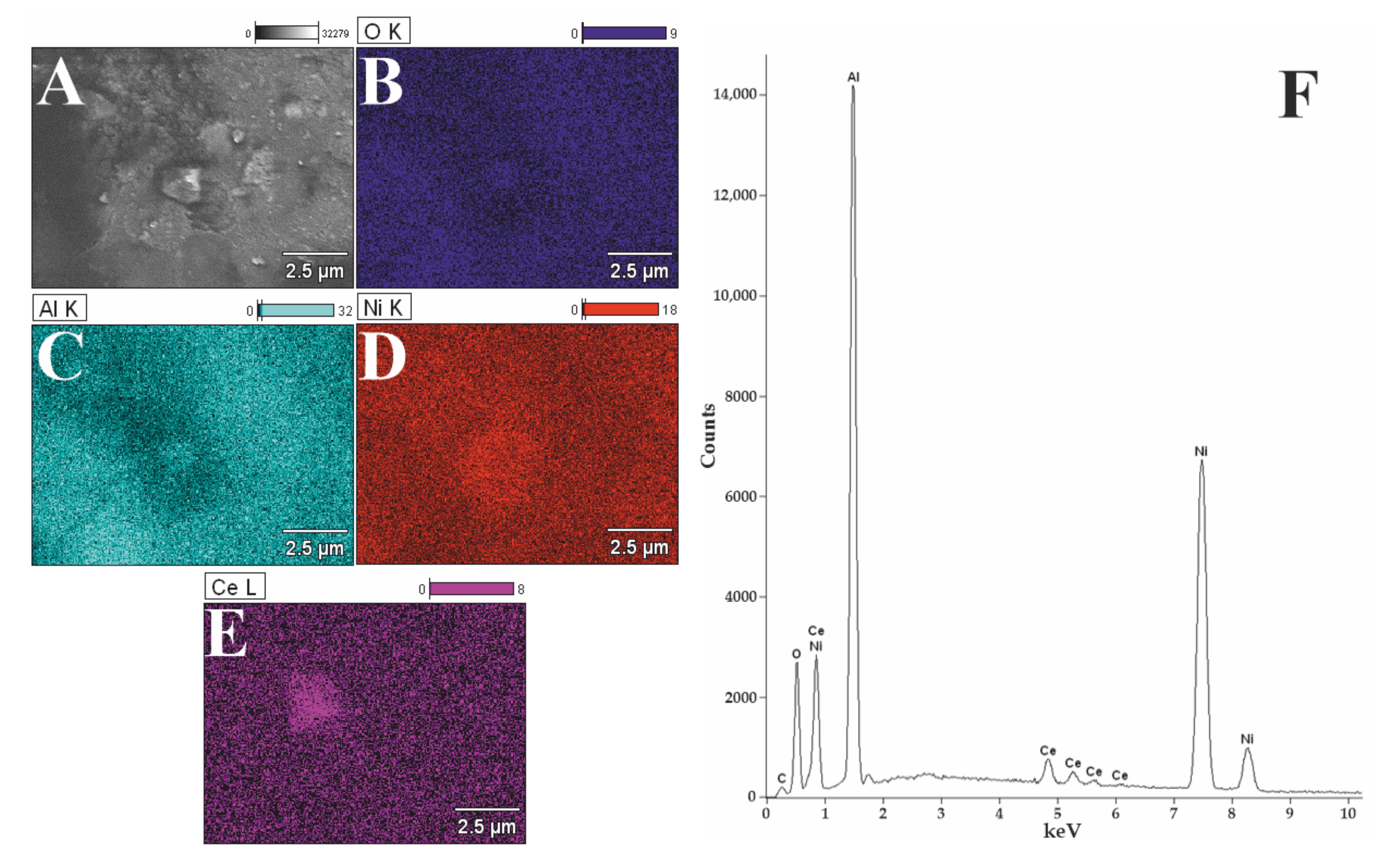
3.2. Morphology Studies
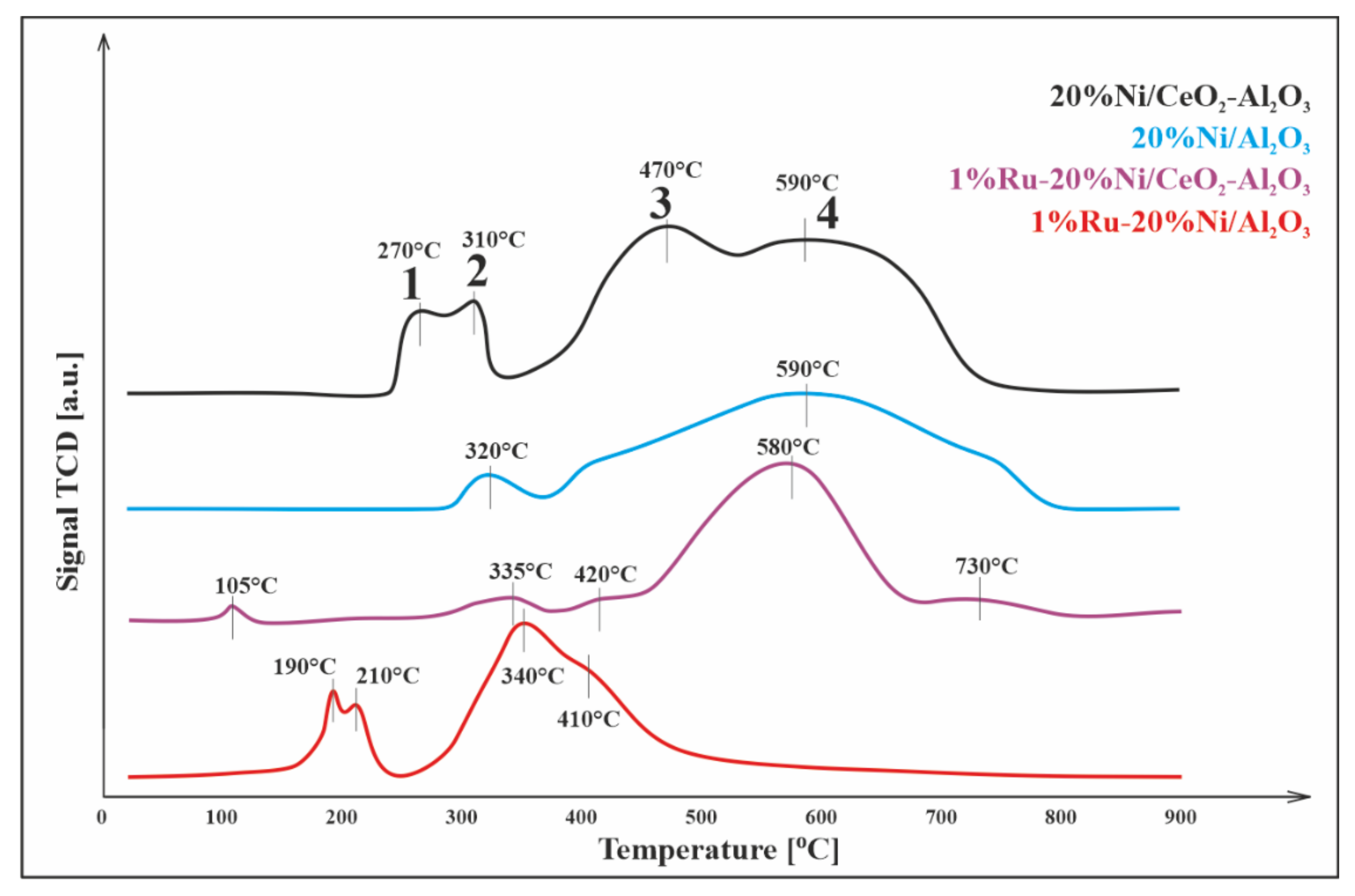
3.3. Reduction Studies (TPR–H2)
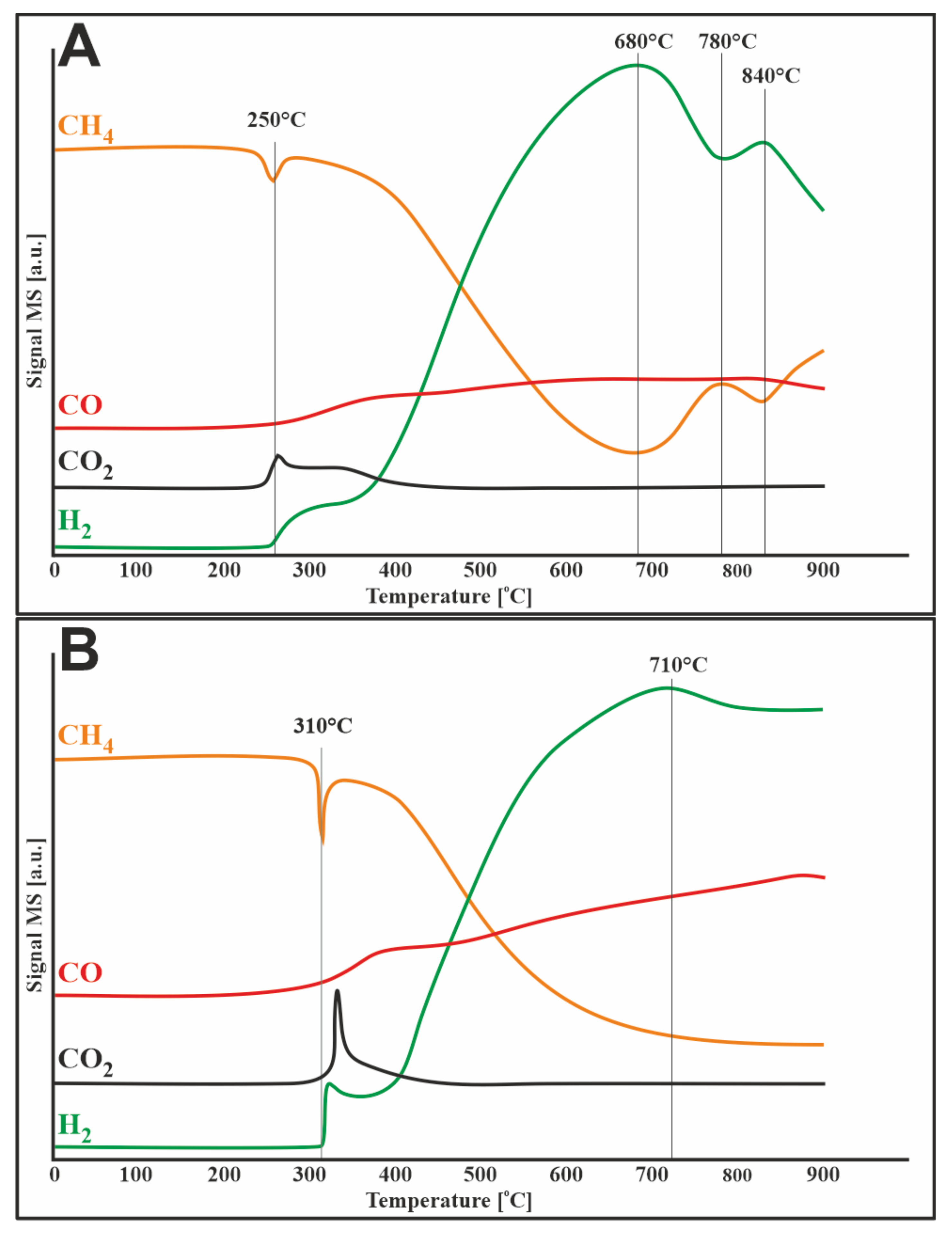
3.4. TPR–CH4 Studies
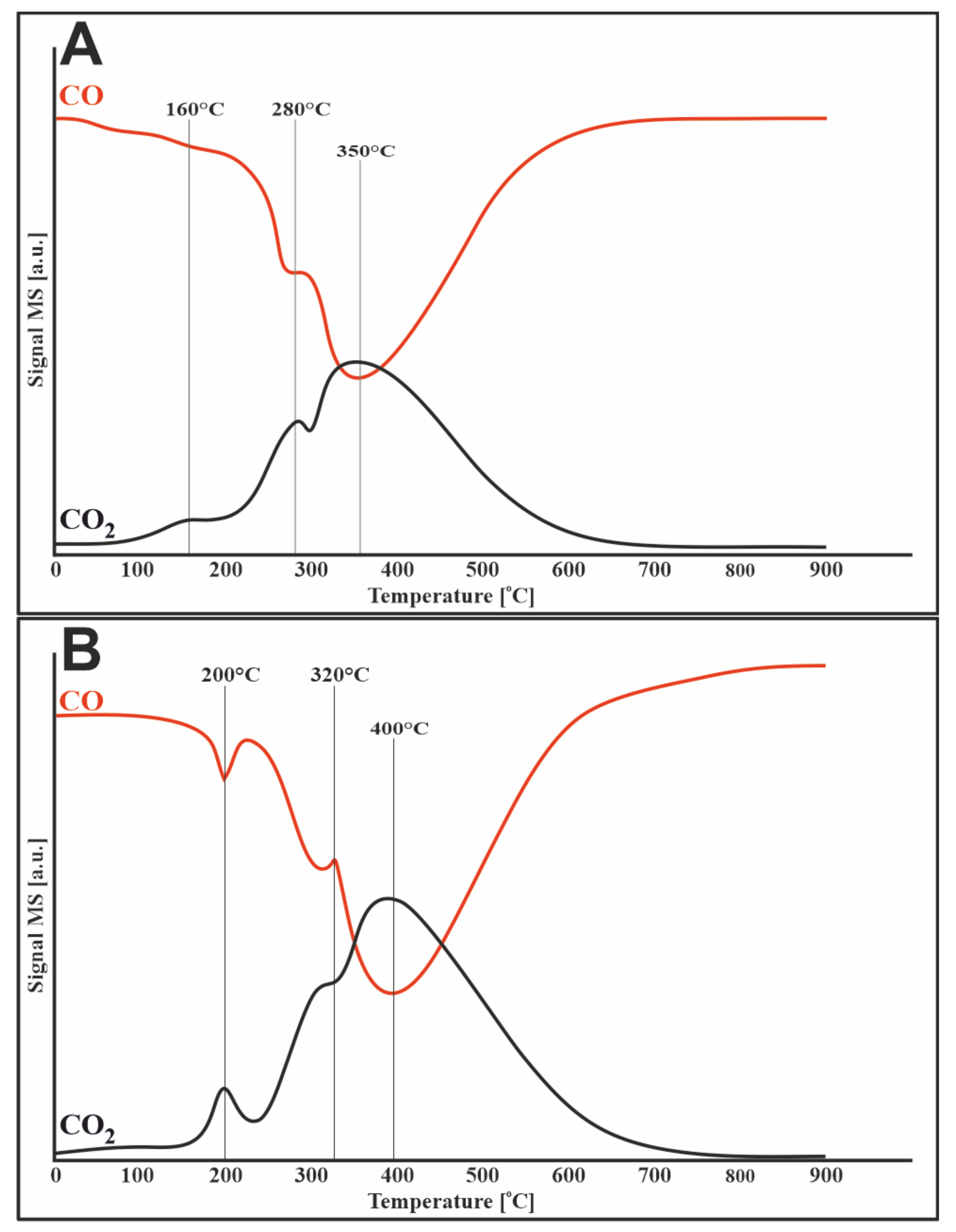
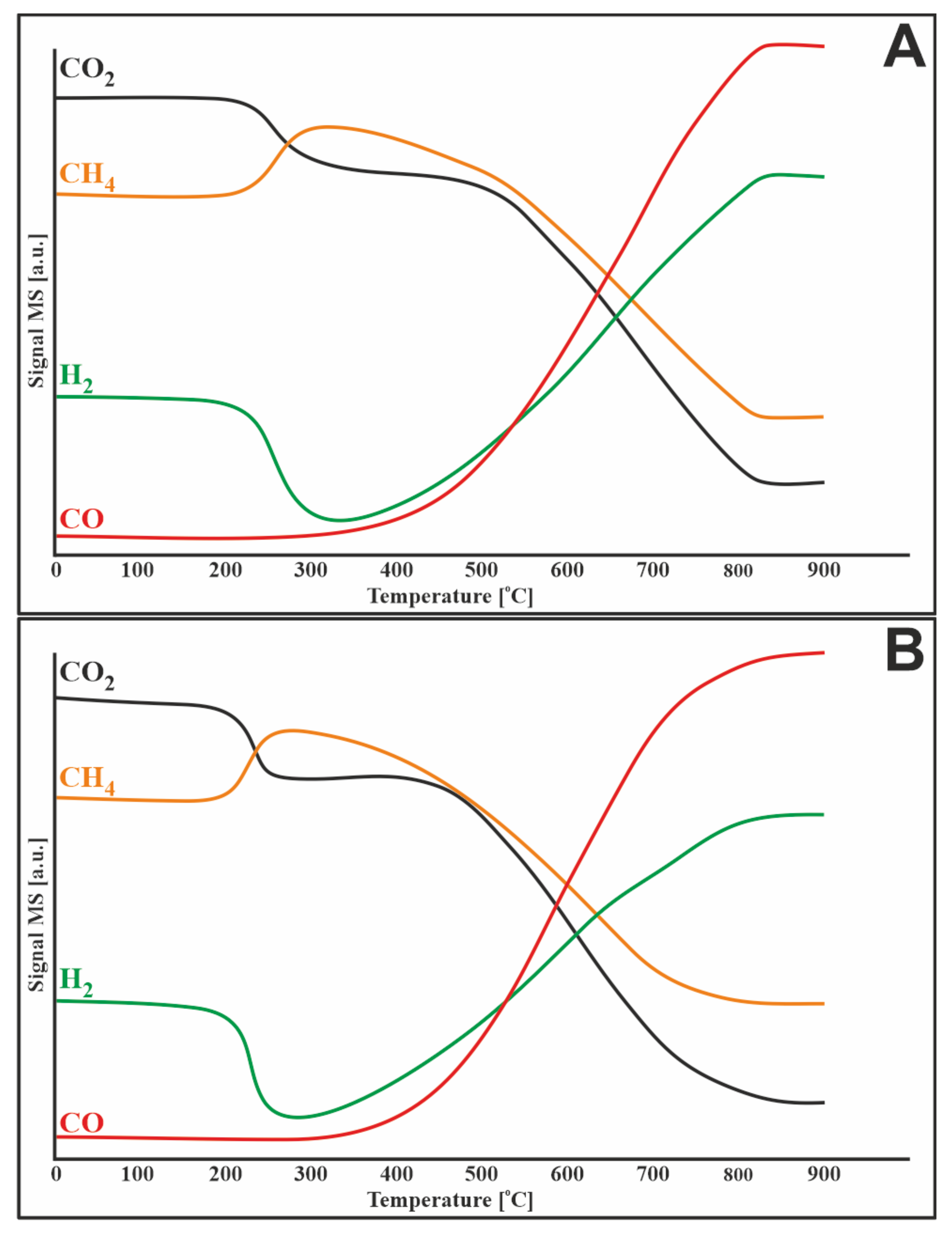
3.5. Methane Decomposition and Carbon Monoxide Disproportionation
3.6. Catalytic Activity Measurements/Total Organic Carbon (TOC) Analysis
3.7. Reactivity of Carbon Deposit towards Different Gasifying Agents (O2, H2, CO2, and H2O)
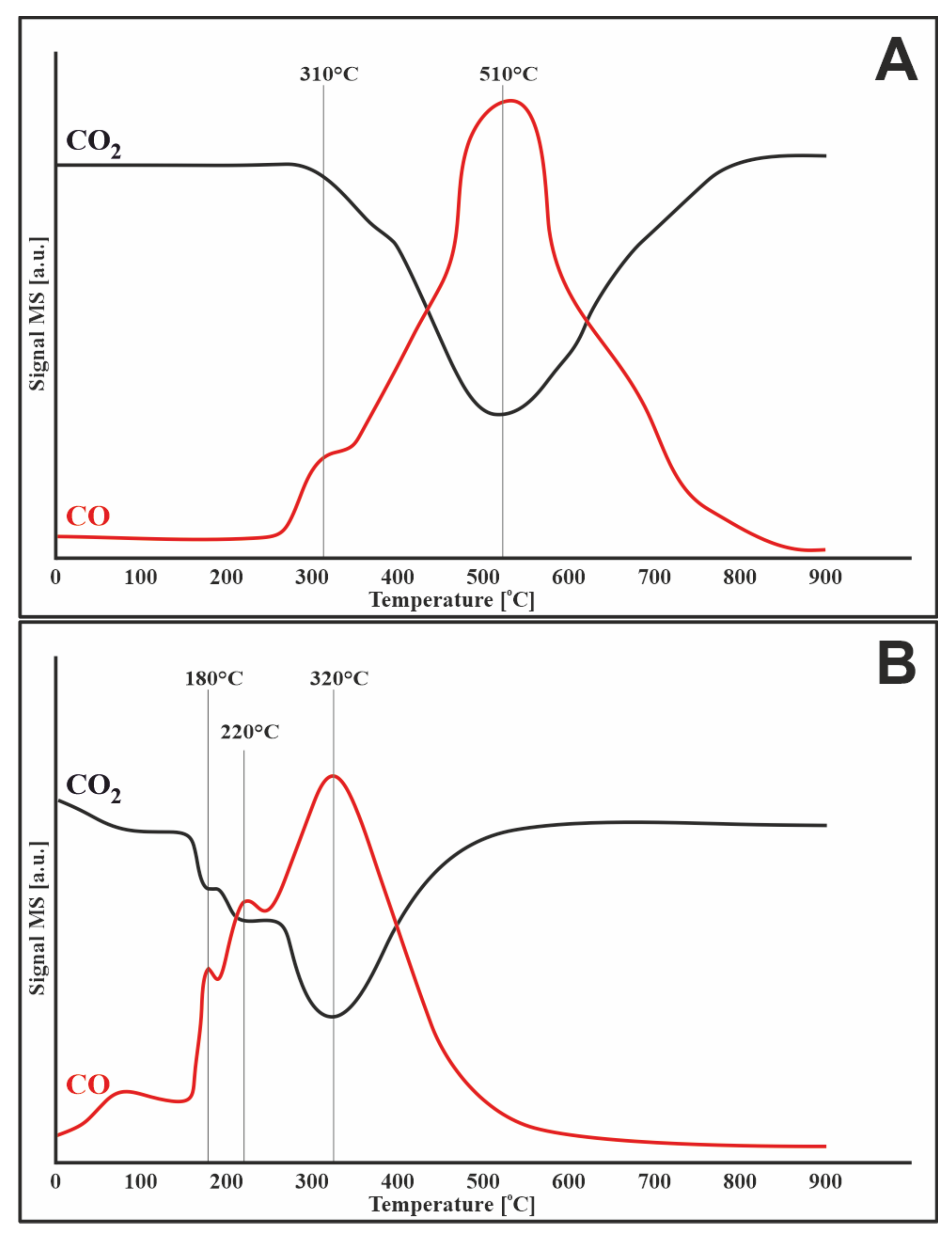
3.7.1. Oxygen
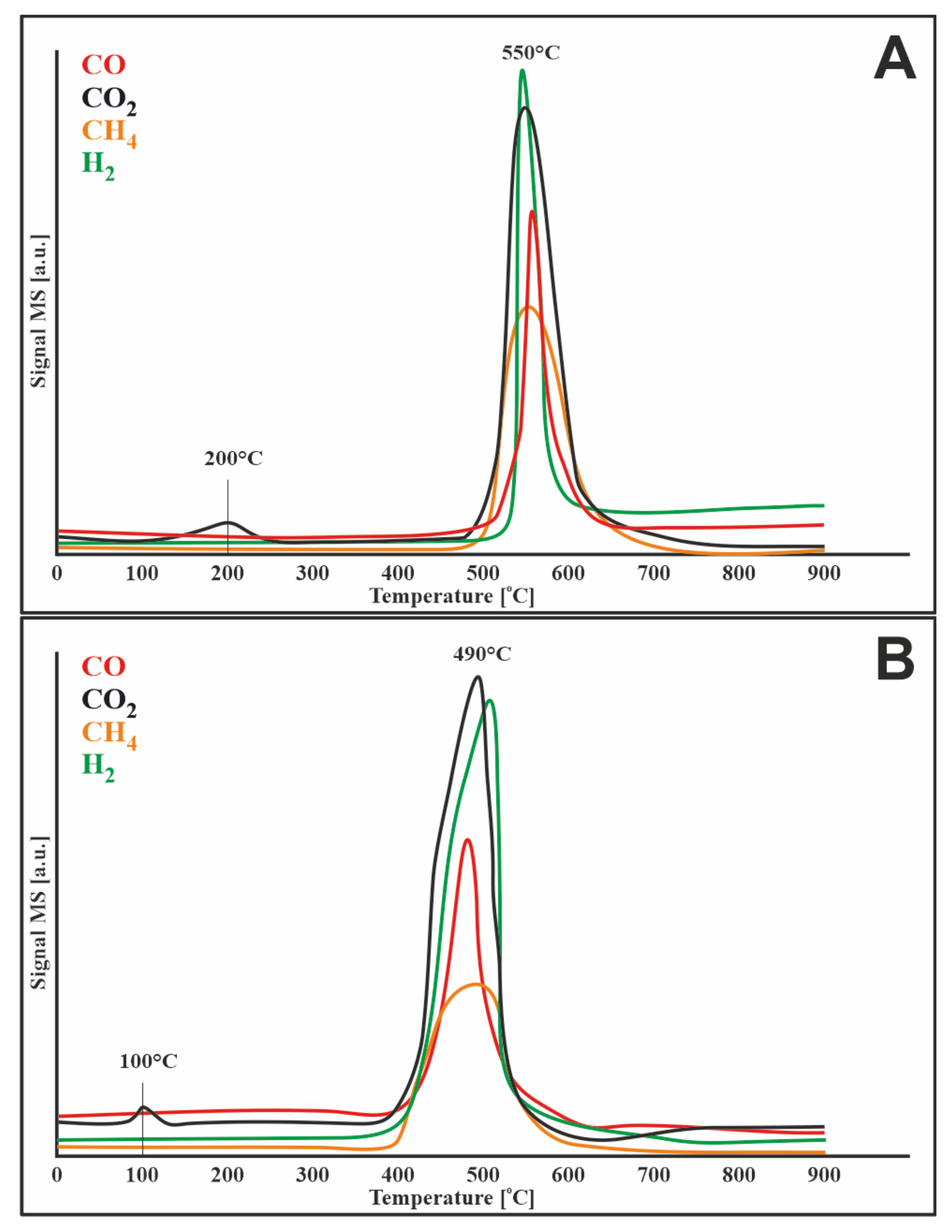
3.7.2. Hydrogen
3.7.3. Carbon Dioxide
3.7.4. Water Vapor
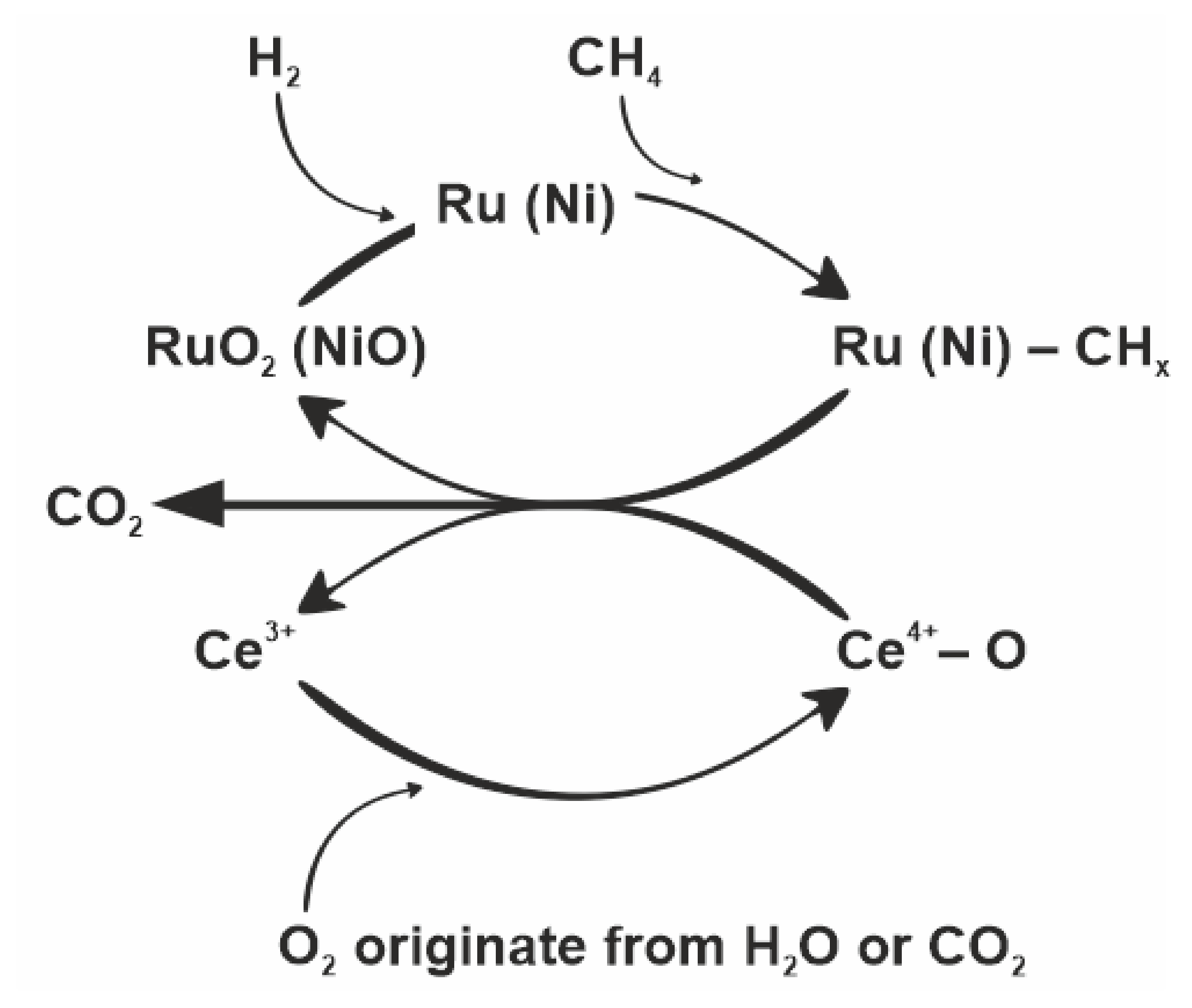
4. Influence of Ruthenium on the MRM Process
- The addition of ruthenium causes the formation of Ru-CHx sites on support. (Equation (15)) [34].
- Oxidation of Ru with activated CO2 results in the formation of Ru-O and Ru-CO species (Equations (16) and (17)). Ru-O can also originate from the dissociation of water vapor (with the formation of hydrogen, Equation (18)). This can take place directly through dissociation on the metal particles (Equation (18)) or indirectly through the support (Equations (19)–(22)) [34].
- Transfer of oxygen from Ru-O to Ru-CHx with the formation of further CO, CO2, and H2 (Equations (23)–(25)). As a consequence, Ru is regenerated (Equations (24) and (25)) [34].
- The adsorbed hydrogen (Ru-H) can also combine with the adsorbed oxygen (Ru-O) to form a hydroxyl group and then combine with the hydrogen to form water vapor (H2O(g)) which then desorbs from the ruthenium surface onto effluent gas mixture (Equations (26) and (27)) [34].
5. Role of Cerium Oxide the MRM Process
6. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mierczynski, P.; Ciesielski, R.; Zakrzewski, M.; Dawid, B.; Mosinska, M.; Maniukiewicz, W.; Dubkov, S.; Gromov, D.; Szynkowska, M.I.; Witonska, I.; et al. Carbon Deposits Formed on the Surface of Ru–Ni Catalysts During the Mixed Reforming of Methane Process. Kinet. Catal. 2018, 59, 372–377. [Google Scholar] [CrossRef]
- Mierczynski, P.; Ciesielski, R.; Zakrzewski, M.; Dawid, B.; Mosinska, M.; Kedziora, A.; Maniukiewicz, W.; Dubkov, S.; Gromov, D.; Szynkowska, M.; et al. Supported Ru−Ni Catalysts for Biogas and Biohydrogen Conversion into Syngas. Kinet. Catal. 2018, 59, 509–513. [Google Scholar] [CrossRef]
- McCarty, J.G.; Wise, H. Hydrogenation of Surface Carbon on Alumina-Supported Nickel. J. Catal. 1979, 57, 406–416. [Google Scholar] [CrossRef]
- Van Beurden, P. On the Catalytic Aspects of Steam-Methane Reforming a Literature Survey. 2004. Available online: https://www.semanticscholar.org/paper/ON-THE-CATALYTIC-ASPECTS-OF-STEAM-METHANE-REFORMING-Beurden/acc982524c316ebee7e538dc508be0792ceafae0 (accessed on 5 May 2021).
- Bartholomew, C.H. Mechanisms of Catalyst Deactivation. Appl. Catal. A Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Trimm, D.L. Control of Coking. Chem. Eng. Process. Process. Intensif. 1984, 18, 137–148. [Google Scholar] [CrossRef]
- Baker, R.T.K.; Kim, M.S.; Chambers, A.; Park, C.; Rodriguez, N.M. The Relationship between metal particle morphology and the structural characteristics of carbon deposits. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 1997; Volume 111, pp. 99–109. [Google Scholar]
- Bernardo, C.A.; Trimm, D.L. The Kinetics of Gasification of Carbon Deposited on Nickel Catalysts. Carbon 1979, 17, 115–120. [Google Scholar] [CrossRef]
- Damyanova, S.; Pawelec, B.; Arishtirova, K.; Huerta, M.V.M.; Fierro, J.L.G. The Effect of CeO2 on the Surface and Catalytic Properties of Pt/CeO2–ZrO2 Catalysts for Methane Dry Reforming. Appl. Catal. B Environ. 2009, 89, 149–159. [Google Scholar] [CrossRef]
- Terribile, D.; Trovarelli, A.; de Leitenburg, C.; Primavera, A.; Dolcetti, G. Catalytic Combustion of Hydrocarbons with Mn and Cu-Doped Ceria–Zirconia Solid Solutions. Catal. Today 1999, 47, 133–140. [Google Scholar] [CrossRef]
- Aneggi, E.; de Leitenburg, C.; Dolcetti, G.; Trovarelli, A. Promotional Effect of Rare Earths and Transition Metals in the Combustion of Diesel Soot over CeO2 and CeO2–ZrO2. Catal. Today 2006, 114, 40–47. [Google Scholar] [CrossRef]
- Kašpar, J.; di Monte, R.; Fornasiero, P.; Graziani, M.; Bradshaw, H.; Norman, C. Dependency of the Oxygen Storage Capacity in Zirconia–Ceria Solid Solutions upon Textural Properties. Top. Catal. 2001, 16, 83–87. [Google Scholar] [CrossRef]
- Crisafulli, C.; Scirè, S.; Minicò, S.; Solarino, L. Ni–Ru Bimetallic Catalysts for the CO2 Reforming of Methane. Appl. Catal. A Gen. 2002, 225, 1–9. [Google Scholar] [CrossRef]
- Jafarian, S.; Tavasoli, A.; Karimi, A.; Norouzi, O. Steam Reforming of Bagasse to Hydrogen and Synthesis Gas Using Ruthenium Promoted Ni Fe/γ Al2O3 Nano-Catalysts. Int. J. Hydrogen Energy 2017, 42, 5505–5512. [Google Scholar] [CrossRef]
- Park, J.-H.; Chang, T.-S. Promotional Effect of Ruthenium Addition to Co/α-Al2O3 Catalyst for Dry Reforming of Methane. Catal. Lett. 2019, 149, 3148–3159. [Google Scholar] [CrossRef]
- Jabbour, K.; el Hassan, N.; Casale, S.; Estephane, J.; el Zakhem, H. Promotional Effect of Ru on the Activity and Stability of Co/SBA-15 Catalysts in Dry Reforming of Methane. Int. J. Hydrogen Energy 2014, 39, 7780–7787. [Google Scholar] [CrossRef]
- Carvalho, L.S.; Martins, A.R.; Reyes, P.; Oportus, M.; Albonoz, A.; Vicentini, V.; do Carmo Rangel, M. Preparation and Characterization of Ru/MgO-Al2O3 Catalysts for Methane Steam Reforming. Catal. Today 2009, 142, 52–60. [Google Scholar] [CrossRef]
- Morales-Cano, F.; Lundegaard, L.F.; Tiruvalam, R.R.; Falsig, H.; Skjøth-Rasmussen, M.S. Improving the Sintering Resistance of Ni/Al2O3 Steam-Reforming Catalysts by Promotion with Noble Metals. Appl. Catal. A Gen. 2015, 498, 117–125. [Google Scholar] [CrossRef]
- Jeong, J.H.; Lee, J.W.; Seo, D.J.; Seo, Y.; Yoon, W.L.; Lee, D.K.; Kim, D.H. Ru-Doped Ni Catalysts Effective for the Steam Reforming of Methane without the Pre-Reduction Treatment with H2. Appl. Catal. A Gen. 2006, 302, 151–156. [Google Scholar] [CrossRef]
- Crisafulli, C.; Scirè, S.; Maggiore, R.; Minicò, S.; Galvagno, S. CO2 Reforming of Methane over Ni-Ru and Ni-Pd Bimetallic Catalysts. Catal. Lett. 1999, 59, 21–26. [Google Scholar] [CrossRef]
- Mierczynski, P.; Mosinska, M.; Maniukiewicz, W.; Vasilev, K.; Szynkowska-Jozwik, M.I. Oxy-Steam Reforming of Liquefied Natural Gas (LNG) on Mono- and Bimetallic (Ag, Pt, Pd or Ru)/Ni Catalysts. Catalysts 2021, 11, 1401. [Google Scholar] [CrossRef]
- Mierczynski, P.; Maniukiewicz, W.; Maniecki, T. Comparative Studies of Pd, Ru, Ni, Cu/ZnAl2O4 Catalysts for the Water Gas Shift Reaction. Open Chem. 2013, 11, 912–919. [Google Scholar] [CrossRef]
- Profeti, L.P.R.; Ticianelli, E.A.; Assaf, E.M. Production of Hydrogen via Steam Reforming of Biofuels on Ni/CeO2–Al2O3 Catalysts Promoted by Noble Metals. Int. J. Hydrogen Energy 2009, 34, 5049–5060. [Google Scholar] [CrossRef]
- Wang, S.; Lu, G.Q.M. Role of CeO2 in Ni/CeO2–Al2O3 Catalysts for Carbon Dioxide Reforming of Methane. Appl. Catal. B Environ. 1998, 19, 267–277. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Baklavaridis, A.; Papadakis, V.G.; Goula, M.A. Synthesis Gas Production via the Biogas Reforming Reaction Over Ni/MgO–Al2O3 and Ni/CaO–Al2O3 Catalysts. Waste Biomass Valorization 2016, 7, 725–736. [Google Scholar] [CrossRef]
- Li, D.; Atake, I.; Shishido, T.; Oumi, Y.; Sano, T.; Takehira, K. Self-Regenerative Activity of Ni/Mg(Al)O Catalysts with Trace Ru during Daily Start-up and Shut-down Operation of CH4 Steam Reforming. J. Catal. 2007, 250, 299–312. [Google Scholar] [CrossRef]
- Nurunnabi, M.; Murata, K.; Okabe, K.; Inaba, M.; Takahara, I. Effect of Mn Addition on Activity and Resistance to Catalyst Deactivation for Fischer–Tropsch Synthesis over Ru/Al2O3 and Ru/SiO2 Catalysts. Catal. Commun. 2007, 8, 1531–1537. [Google Scholar] [CrossRef]
- Jiang, P.; Shang, Y.; Cheng, T.; Bi, Y.; Shi, K.; Wei, S.; Xu, G.; Zhen, K. Methane Decomposition Over Ni/α-Al2O3 Promoted by La2O3 and CeO2. J. Nat. Gas Chem. 2003, 12, 183–188. [Google Scholar] [CrossRef]
- Luisetto, I.; Sarno, C.; de Felicis, D.; Basoli, F.; Battocchio, C.; Tuti, S.; Licoccia, S.; di Bartolomeo, E. Ni Supported on γ-Al2O3 Promoted by Ru for the Dry Reforming of Methane in Packed and Monolithic Reactors. Fuel Process. Technol. 2017, 158, 130–140. [Google Scholar] [CrossRef]
- Wu, H.C.; Chang, Y.C.; Wu, J.H.; Lin, J.H.; Lin, I.K.; Chen, C.S. Methanation of CO 2 and Reverse Water Gas Shift Reactions on Ni/SiO 2 Catalysts: The Influence of Particle Size on Selectivity and Reaction Pathway. Catal. Sci. Technol. 2015, 5, 4154–4163. [Google Scholar] [CrossRef]
- Shtyka, O.; Zakrzewski, M.; Ciesielski, R.; Kedziora, A.; Dubkov, S.; Ryazanov, R.; Szynkowska, M.; Maniecki, T. Efficient Removal of the Carbon Deposits Formed during the Mixed Methane Reforming over Ni/Al2O3. Korean J. Chem. Eng. 2020, 37, 209–215. [Google Scholar] [CrossRef]
- Yang, Y.; Li, W.; Xu, H. A New Explanation for the Carbon Deposition and Elimination over Supported Ni, Ni-Ce and Ni-Co Catalysts for CO2-Reforming of Methane. React. Kinet. Catal. Lett. 2002, 77, 155–162. [Google Scholar] [CrossRef]
- Walker, P.L.; Rusinko, F.; Austin, L.G. Gas Reactions of Carbon. Adv. Catal. 1959, 11, 133–221. [Google Scholar]
- Matsui, N.; Anzai, K.; Akamatsu, N.; Nakagawa, K.; Ikenaga, N.; Suzuki, T. Reaction Mechanisms of Carbon Dioxide Reforming of Methane with Ru-Loaded Lanthanum Oxide Catalyst. Appl. Catal. A Gen. 1999, 179, 247–256. [Google Scholar] [CrossRef]
- Kurungot, S.; Yamaguchi, T. Stability Improvement of Rh/γ-Al2O3 Catalyst Layer by Ceria Doping for Steam Reforming in an Integrated Catalytic Membrane Reactor System. Catal. Lett. 2004, 92, 181–187. [Google Scholar] [CrossRef]
- Dong, W. Methane Reforming over Ni/Ce-ZrO2 Catalysts: Effect of Nickel Content. Appl. Catal. A Gen. 2002, 226, 63–72. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Metal Content in the Catalyst | Average Size of Ni Crystallites (nm) | Specific Surface Area (m2/g) | Pore Size (nm) |
|---|---|---|---|---|
| 5%CeO2-95%Al2O3 | - | - | 170 | 1.9–2.3 |
| 20%Ni/5%CeO2-95%Al2O3 | 18.2% Ni | 45 | 147 | 2.6–3.6 |
| 1%Ru-20%Ni/5%CeO2-95%Al2O3 | 18.8% Ni | 16 | 149 | 2.5–3.6 |
| Catalytic System/Sample Carbonization Temperature (°C) | Conversion after 3 h Reaction | Carbon Content (%) after: | |||||
|---|---|---|---|---|---|---|---|
| CH4 (%) | CO2 (%) | MRM Process | TPSR (5%H2-95%Ar) | TPSR (99.99%H2) | TPSR (5%CO2-95%Ar) | TPSR (5%H2O-95%Ar) | |
| 20%Ni/CeO2-Al2O3 [650] | 44 | 44 | 2.3 | 0 | 0 | 0 | 0 |
| 20%Ni/CeO2-Al2O3 [700] | 68 | 62 | 4.6 | 0 | 0 | 0 | 0 |
| 20%Ni/CeO2-Al2O3 [750] | 78 | 71 | 3.5 | 0 | 0 | 0 | 0 |
| 1%Ru-20%Ni/CeO2-Al2O3 [650] | 57 | 54 | 1.7 | 0 | 0 | 0 | 0 |
| 1%Ru-20%Ni/CeO2-Al2O3 [700] | 88 | 84 | 3.5 | 0 | 0 | 0 | 0 |
| 1%Ru-20%Ni/CeO2-Al2O3 [750] | 93 | 86 | 1.5 | 0 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakrzewski, M.; Shtyka, O.; Ciesielski, R.; Kedziora, A.; Maniukiewicz, W.; Arcab, N.; Maniecki, T. Effect of Ruthenium and Cerium Oxide (IV) Promotors on the Removal of Carbon Deposit Formed during the Mixed Methane Reforming Process. Materials 2021, 14, 7581. https://doi.org/10.3390/ma14247581
Zakrzewski M, Shtyka O, Ciesielski R, Kedziora A, Maniukiewicz W, Arcab N, Maniecki T. Effect of Ruthenium and Cerium Oxide (IV) Promotors on the Removal of Carbon Deposit Formed during the Mixed Methane Reforming Process. Materials. 2021; 14(24):7581. https://doi.org/10.3390/ma14247581
Chicago/Turabian StyleZakrzewski, Mateusz, Oleksandr Shtyka, Radoslaw Ciesielski, Adam Kedziora, Waldemar Maniukiewicz, Natalia Arcab, and Tomasz Maniecki. 2021. "Effect of Ruthenium and Cerium Oxide (IV) Promotors on the Removal of Carbon Deposit Formed during the Mixed Methane Reforming Process" Materials 14, no. 24: 7581. https://doi.org/10.3390/ma14247581
APA StyleZakrzewski, M., Shtyka, O., Ciesielski, R., Kedziora, A., Maniukiewicz, W., Arcab, N., & Maniecki, T. (2021). Effect of Ruthenium and Cerium Oxide (IV) Promotors on the Removal of Carbon Deposit Formed during the Mixed Methane Reforming Process. Materials, 14(24), 7581. https://doi.org/10.3390/ma14247581