
Formation of Citrazinic Acid Ions and Their Contribution to Optical and Magnetic Features of Carbon Nanodots: A Combined Experimental and Computational Approach

 , , ,
, , ,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Thermodynamic Cycles
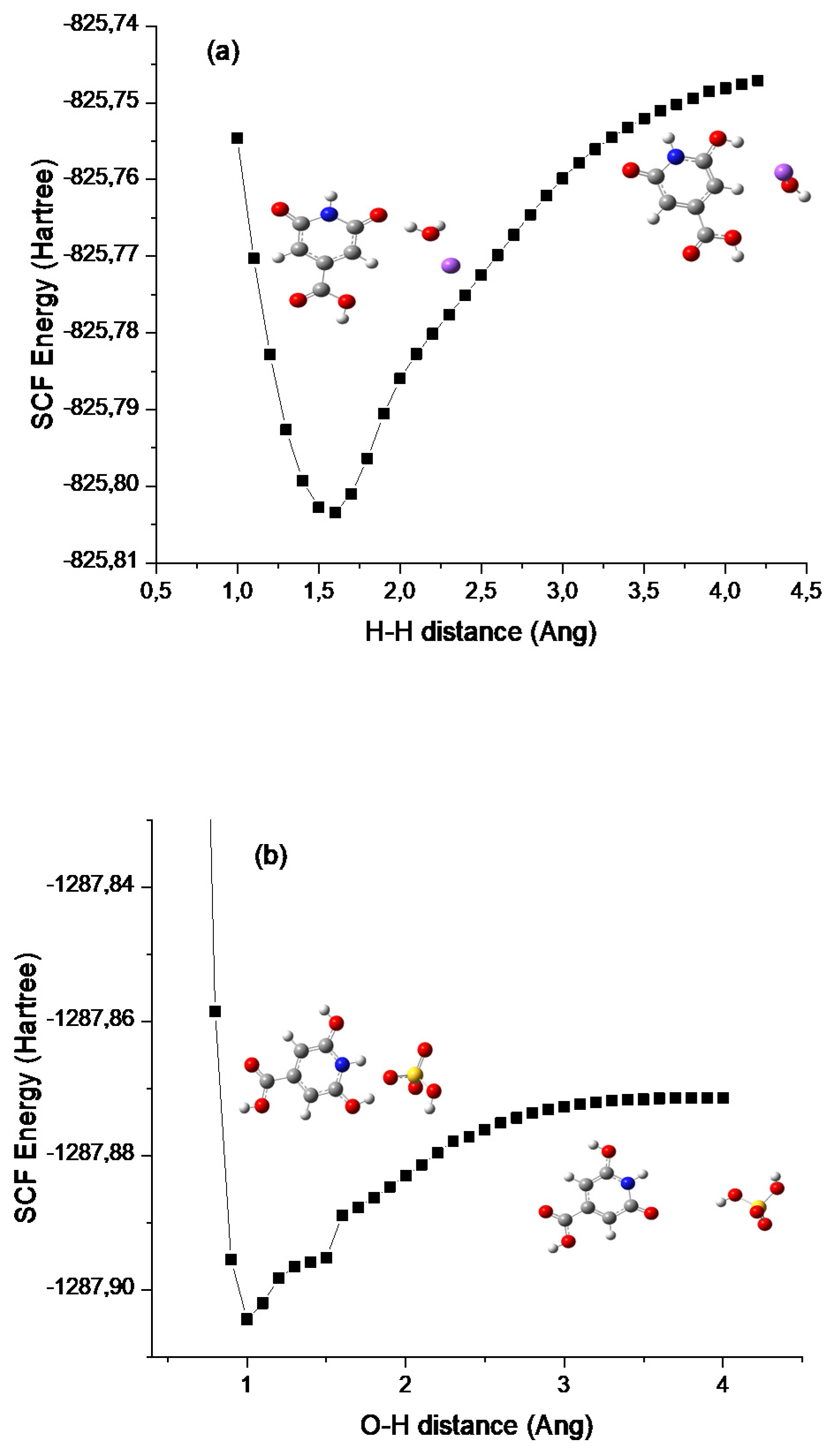
3.2. Potential Energy Surface (PES) Simulations
3.3. Optical Properties
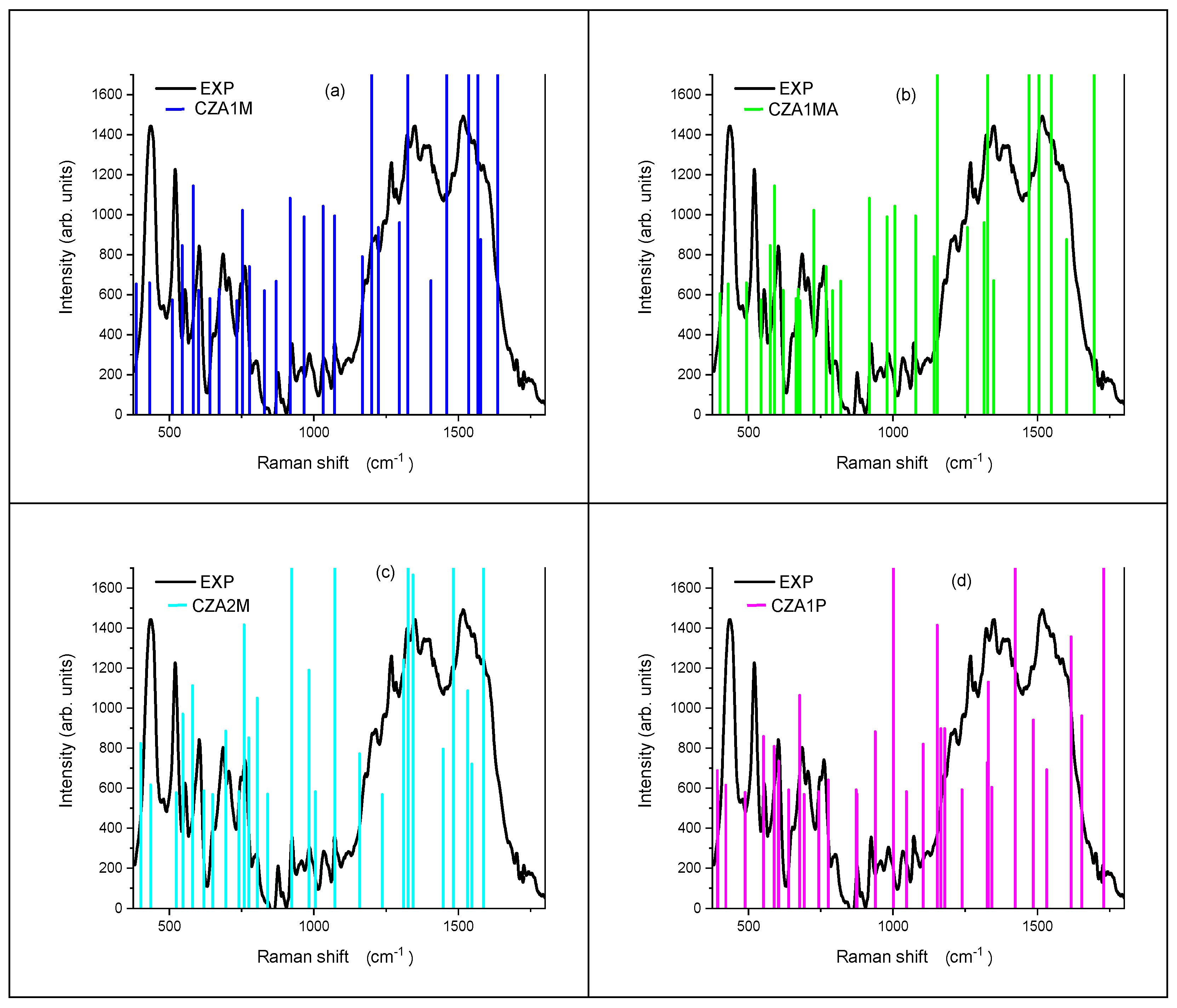
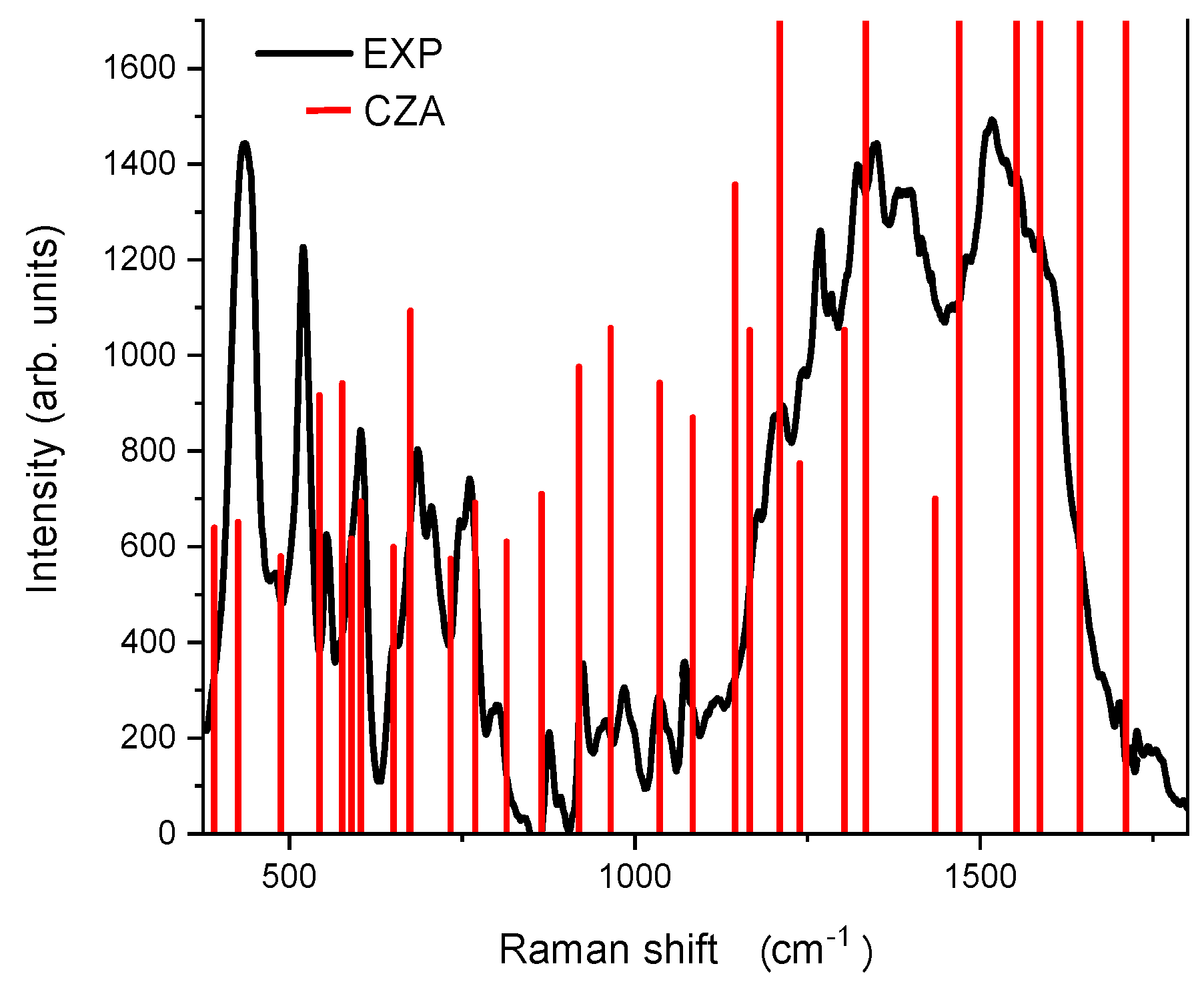
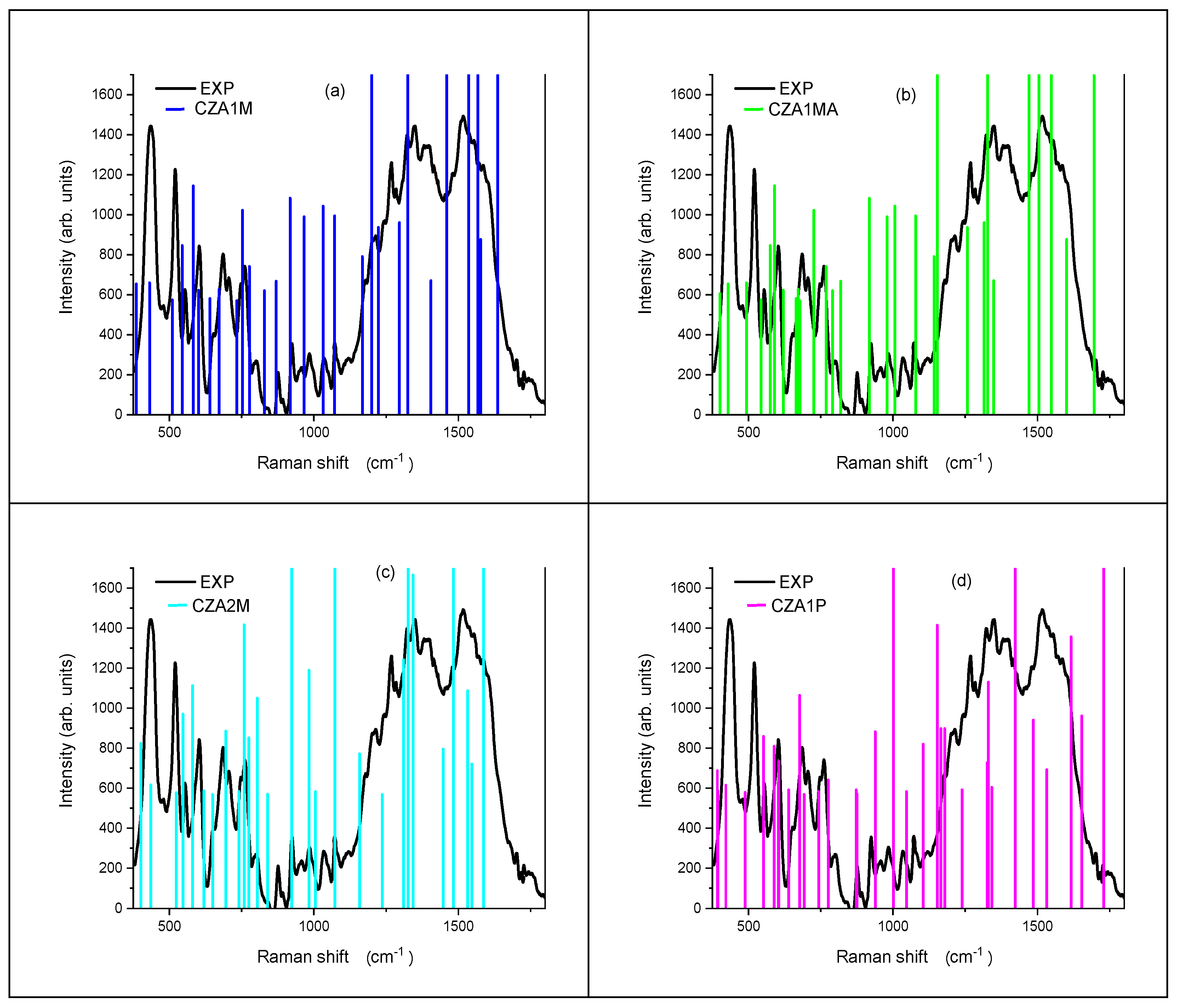
3.4. Vibrational Analysis
3.5. NMR Calculations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reckmeier, C.J.; Schneider, J.; Susha, A.S.; Rogach, A.L. Luminescent colloidal carbon dots: Optical properties and effects of doping [Invited]. Opt. Express 2016, 24, A312–A340. [Google Scholar] [CrossRef]
- Mintz, K.J.; Zhou, Y.; Leblanc, R.M. Recent development of carbon quantum dots regarding their optical properties, photoluminescence mechanism, and core structure. Nanoscale 2019, 11, 4634–4652. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Meng, T.; He, P.; Shi, Y.; Li, Y.; Li, X.; Fan, L.; Yang, S. Carbon quantum dots: An emerging material for optoelectronic applications. J. Mater. Chem. C 2019, 7, 6820–6835. [Google Scholar] [CrossRef]
- Carbonaro, C.M.; Corpino, R.; Salis, M.; Mocci, F.; Thakkar, S.V.; Olla, C.; Ricci, P.C. On the emission properties of carbon dots: Reviewing data and discussing models. C J. Carbon Res. 2019, 5, 60. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Gadly, T.; Neogy, S.; Ghosh, S.K.; Kumbhakar, M. Molecular origin and self-assembly of fluorescent carbon nanodots in polar solvents. J. Phys. Chem. Lett. 2017, 8, 1044–1052. [Google Scholar] [CrossRef]
- Carbonaro, C.M.; Chiriu, D.; Stagi, L.; Casula, M.F.; Thakkar, S.V.; Malfatti, L.; Suzuki, K.; Ricci, P.C.; Corpino, R. Carbon dots in water and mesoporous matrix: Chasing the origin of their photoluminescence. J. Phys. Chem. C 2018, 122, 25638–25650. [Google Scholar] [CrossRef]
- Shamsipur, M.; Barati, A.; Taherpour, A.A.; Jamshidi, M. Resolving the multiple emission centers in carbon Dots: From fluorophore molecular states to aromatic domain states and carbon-core states. J. Phys. Chem. Lett. 2018, 9, 4189–4198. [Google Scholar] [CrossRef]
- Ehrat, F.; Bhattacharyya, S.; Schneider, J.; Löf, A.; Wyrwich, R.; Rogach, A.L.; Stolarczyk, J.K.; Urban, A.S.; Feldmann, J. Tracking the source of carbon dot photoluminescence: Aromatic domains versus molecular fluorophores. Nano Lett. 2017, 17, 7710–7716. [Google Scholar] [CrossRef]
- Xiong, Y.; Schneider, J.; Reckmeier, C.J.; Huang, H.; Kasák, P.; Rogach, A.L. Carbonization conditions influence the emission characteristics and the stability against photobleaching of nitrogen doped carbon dots. Nanoscale 2017, 9, 11730–11738. [Google Scholar] [CrossRef] [PubMed]
- Righetto, M.; Privitera, A.; Fortunati, I.; Mosconi, D.; Zerbetto, M.; Curri, M.L.; Corricelli, M.; Moretto, A.; Agnoli, S.; Franco, L.; et al. Spectroscopic insights into carbon dot systems. J. Phys. Chem. Lett. 2017, 8, 2236–2242. [Google Scholar] [CrossRef]
- Wang, T.; Wang, A.; Wang, R.; Liu, Z.; Sun, Y.; Shan, G.; Chen, Y.; Liu, Y. Carbon dots with molecular fluorescence and their application as a “turn-off” fluorescent probe for ferricyanide detection. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Schneider, J.; Ushakova, E.V.; Rogach, A.L. Influence of molecular fluorophores on the research field of chemically synthesized carbon dots. Nano Today 2018, 23, 124–139. [Google Scholar] [CrossRef]
- Kasprzyk, W.; Świergosz, T.; Bednarz, S.; Walas, K.; Bashmakova, N.V.; Bogdał, D. Luminescence phenomena of carbon dots derived from citric acid and urea-a molecular insight. Nanoscale 2018, 10, 13889–13894. [Google Scholar] [CrossRef]
- Reckmeier, C.J.; Schneider, J.; Xiong, Y.; Häusler, J.; Kasák, P.; Schnick, W.; Rogach, A.L. Aggregated Molecular fluorophores in the ammonothermal synthesis of carbon dots. Chem. Mater. 2017, 29, 10352–10361. [Google Scholar] [CrossRef] [Green Version]
- Holá, K.; Sudolská, M.; Kalytchuk, S.; Nachtigallová, D.; Rogach, A.L.; Otyepka, M.; Zbořil, R. Graphitic Nitrogen triggers red fluorescence in carbon dots. ACS Nano 2017, 11, 12402–12410. [Google Scholar] [CrossRef] [PubMed]
- Sudolská, M.; Otyepka, M. Exact roles of individual chemical forms of nitrogen in the photoluminescent properties of nitrogen-doped carbon dots. Appl. Mater. Today 2017, 7, 190–200. [Google Scholar] [CrossRef]
- Nandy, A.; Kumar, A.; Dwivedi, S.; Pal, S.K.; Panda, D. Connecting the dots of carbon nanodots: Excitation (in)dependency and white-light emission in one-step. J. Phys. Chem. C 2019, 123, 20502–20511. [Google Scholar] [CrossRef]
- Siddique, F.; Langer, M.; Paloncýová, M.; Medved’, M.; Otyepka, M.; Nachtigallová, D.; Lischka, H.; Aquino, A.J.A. Conformational behavior and optical properties of a fluorophore dimer as a model of luminescent centers in carbon dots. J. Phys. Chem. C 2020, 124, 14327–14337. [Google Scholar] [CrossRef]
- Ludmerczki, R.; Mura, S.; Carbonaro, C.M.; Mandity, I.M.; Carraro, M.; Senes, N.; Garroni, S.; Granozzi, G.; Calvillo, L.; Marras, S.; et al. Carbon dots from citric acid and its intermediates formed by thermal decomposition. Chem. Eur. J. 2019, 25, 11963–11974. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Reckmeier, C.J.; Xiong, Y.; Von Seckendorff, M.; Susha, A.S.; Kasak, P.; Rogach, A.L. Molecular fluorescence in citric acid-based carbon dots. J. Phys. Chem. C 2017, 121, 2014–2022. [Google Scholar] [CrossRef]
- Song, Y.; Zhu, S.; Zhang, S.; Fu, Y.; Wang, L.; Zhao, X.; Yang, B. Investigation from chemical structure to photoluminescent mechanism: A type of carbon dots from the pyrolysis of citric acid and an amine. J. Mater. Chem. C 2015, 3, 5976–5984. [Google Scholar] [CrossRef]
- Deeney, C.; Wang, S.; Belhout, S.A.; Gowen, A.; Rodriguez, B.J.; Redmond, G.; Quinn, S.J. Templated microwave synthesis of luminescent carbon nanofibers. RSC Adv. 2018, 8, 12907–12917. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, B.; Embrechts, H.; Damm, C.; Cadranel, A.; Strauss, V.; Distaso, M.; Hinterberger, V.; Guldi, D.M.; Peukert, W. Shedding light on the effective fluorophore structure of high fluorescence quantum yield carbon nanodots. RSC Adv. 2017, 7, 24771–24780. [Google Scholar] [CrossRef] [Green Version]
- Meierhofer, F.; Dissinger, F.; Weigert, F.; Jungclaus, J.; Müller-Caspary, K.; Waldvogel, S.R.; Resch-Genger, U.; Voss, T. Citric acid based carbon dots with amine type stabilizers: pH-specific luminescence and quantum yield characteristics. J. Phys. Chem. C 2020, 124, 8894–8904. [Google Scholar] [CrossRef]
- Strauss, V.; Wang, H.; Delacroix, S.; Ledendecker, M.; Wessig, P. Carbon nanodots revised: The thermal citric acid/urea reaction. Chem. Sci. 2020, 11, 8256–8266. [Google Scholar] [CrossRef]
- Das, A.; Gude, V.; Roy, D.; Chatterjee, T.; De, C.K.; Mandal, P.K. On the molecular origin of photoluminescence of nonblinking carbon dot. J. Phys. Chem. C 2017, 121, 9634–9641. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, Y.; Yu, B.; Wu, Y.; Yu, X.; Guo, S.; Han, B.; Liu, Z. Visible light-driven photoreduction of CO2 to CH4 over TiO2 using a multiple-site ionic liquid as an absorbent and photosensitizer. ACS Sustain. Chem. Eng. 2020, 8, 9088–9094. [Google Scholar] [CrossRef]
- Youk, S.; Hofmann, J.P.; Badamdorj, B.; Völkel, A.; Antonietti, M.; Oschatz, M. Controlling pore size and pore functionality in sp2-conjugated microporous materials by precursor chemistry and salt templating. J. Mater. Chem. A 2020, 8, 21680–21689. [Google Scholar] [CrossRef]
- Masoud, M.S.; Ibrahim, A.A.; Khalil, E.A.; El-Marghany, A. Spectral properties of some metal complexes derived from uracil-thiouracil and citrazinic acid compounds. Spectrochimica Acta Part A Mol. Biomol. Spectrosc. 2007, 67, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Mura, S.; Stagi, L.; Malfatti, L.; Carbonaro, C.M.; Ludmerczki, R.; Innocenzi, P. Modulating the optical properties of citrazinic acid through the monomer-to-dimer transformation. J. Phys. Chem. A 2020, 124, 197–203. [Google Scholar] [CrossRef]
- Stagi, L.; Mura, S.; Malfatti, L.; Carbonaro, C.M.; Ricci, P.C.; Porcu, S.; Secci, F.; Innocenzi, P. Anomalous optical properties of citrazinic acid under extreme pH conditions. ACS Omega 2020, 5, 10958–10964. [Google Scholar] [CrossRef]
- Sarkar, S.; Chowdhury, J.; Dutta, S.; Pal, T. A pH dependent Raman and surface enhanced Raman spectroscopic studies of citrazinic acid aided by theoretical calculations. Spectrochimica Acta Part A Mol. Biomol. Spectrosc. 2016, 169, 108–115. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP density functional methods for a large set of organic molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Cheeseman, J.R. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Liptak, M.D.; Shields, G.C. Accurate pKa calculations for carboxylic acids using complete basis set and Gaussian-n models combined with CPCM continuum solvation methods. J. Am. Chem. Soc. 2001, 123, 7314–7319. [Google Scholar] [CrossRef]
- Pliego, J.R. Thermodynamic cycles and the calculation of pKa. Chem. Phys. Lett. 2003, 367, 145–149. [Google Scholar] [CrossRef]
- Palascak, M.W.; Shields, G.C. Accurate experimental values for the free energies of hydration of H+, OH−, and H3O+. J. Phys. Chem. A 2004, 108, 3692–3694. [Google Scholar] [CrossRef]
- Osaki, T.; Soejima, E. Quadratic scaling functions for obtaining normal vibrational wavenumbers from the B3LYP calculation. Bull. Cent. Res. Inst. Fukuoka Univ. 2010, 42, 129–134. [Google Scholar]
- Cappai, A.; Melis, C.; Stagi, L.; Ricci, P.C.; Mocci, F.; Carbonaro, C.M. Insight the molecular model in carbon dots through experimental and theoretical analysis of citrazinic acid in aqueous solution. J. Chem. Phys. C 2021. [Google Scholar] [CrossRef]




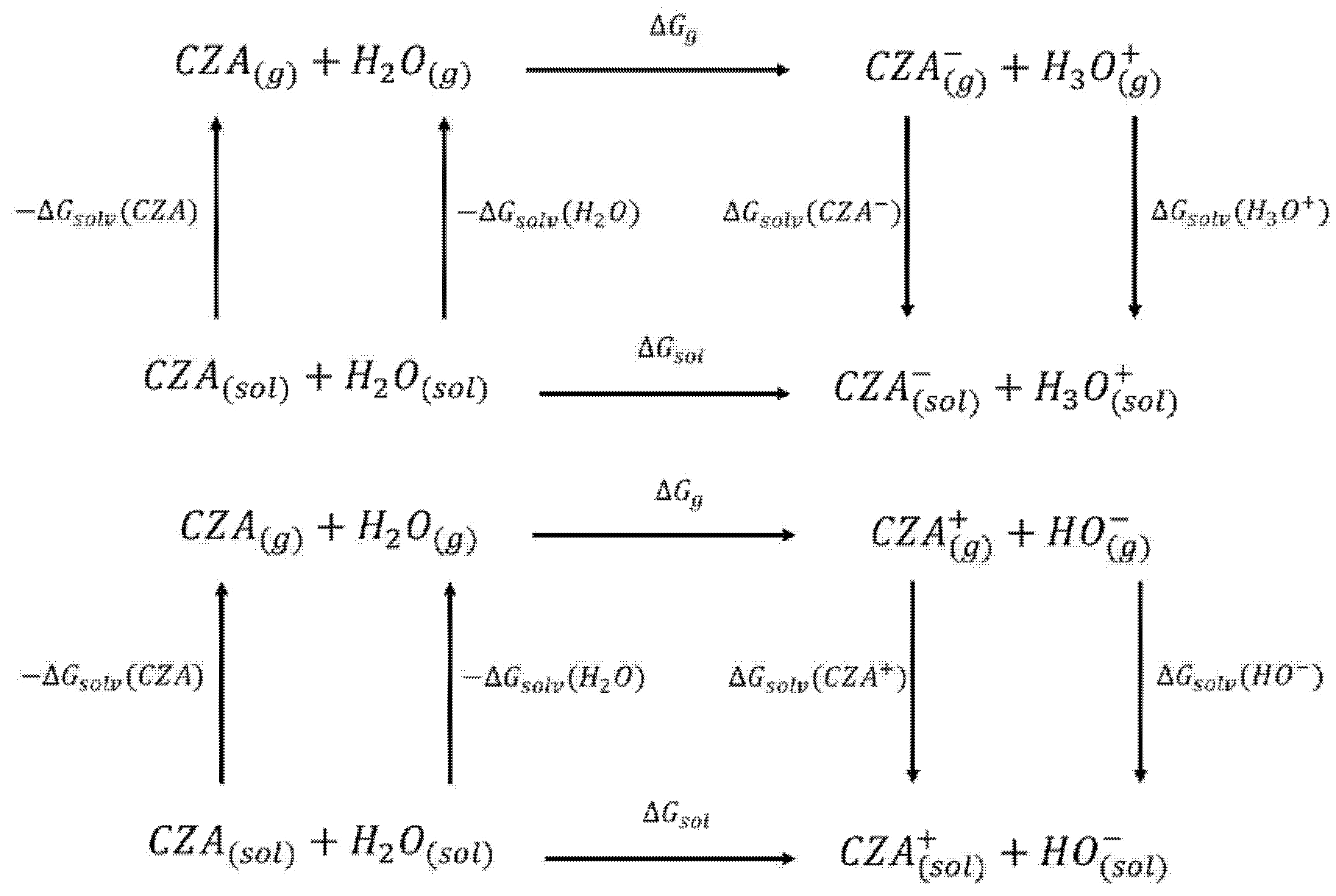{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | q | Structure | ΔGsol (kcal/mol) | pKa |
|---|---|---|---|---|
| CZA | 0 | 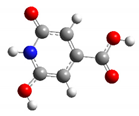 | 0 | - |
| CZA1M | −1 | 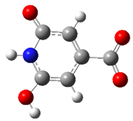 | 18.2 | 7.1 |
| CZA1MA | −1 | 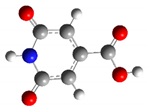 | 13.4 | 3.5 |
| CZA2M | −2 | 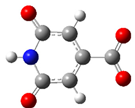 | 37.2 | 21.0 |
| CZA3M | −3 | 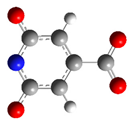 | 91.4 | 60.7 |
| CZA1P | +1 | 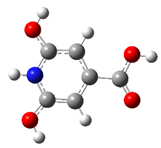 | 31.5 | 16.8 |
| CZA2P | +2 |  | 87.7 | 88.0 |
| Species | HOMO-LUMO (nm) | Oscillator Strength |
|---|---|---|
| CZA CZA1M CZA1MA CZA2M CZA1P | 347.4 307.4 410.5 340.1 305.5 | 0.1197 0.1374 0.1201 0.1738 0.1566 |
| CZA—DMSO CZA—DMF CZA—EtOH | 351.0 351.1 350.4 | 0.1296 0.1303 0.1260 |
| Experimental (cm−1) | Calculated (cm−1) | |
|---|---|---|
| 438 | 425 |  |
| 522 | 487 | 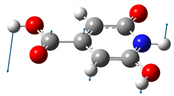 |
| 522 | 543 |  |
| 602 | 590 | 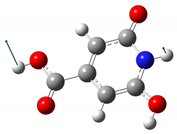 |
| 602 | 603 | 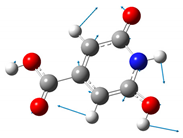 |
| C-Type | CZA | CZA1M | CZA1MA | Exp |
|---|---|---|---|---|
| C-carbonyl/hydroxyl C-β-carbonyl/hydroxyl C-γ C-carboxyl | 159.4 (166.5) 98.8 (105.9) 142.7 (149.7) 166.6 (173.6) | 158.2 (166.2) 96.3 (104.2) 155.5 (163.4) 166.6 (174.5) | 162.4 (173.2) 88.7 (99.4) 137.9 (146.7) 166.6 (177.4) | 162.0 [25] 98.6 [25] 145.1 [25] 166.6 [25] |
| H-Type | CZA | CZA1M | CZA1MA | Exp |
| Aromatic-H Hetero-H | 6.5 7.2 | 6.5 6.5 | 5.7 6.9 | 6.2 [31] 12.1 [31] |
| Dimer Type | Aromatic-H | Hetero-H | |
|---|---|---|---|
| Head-to-Head | 6.5 ppm | 8.1 ppm | 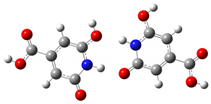 |
| Tail-to-Tail | 6.5 ppm | 8.9 ppm | 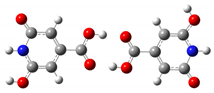 |
| Head-to-Tail | 6.6 ppm | 9.1 ppm | 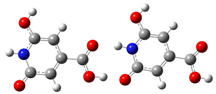 |
| H-Bonding Group | Chem. Shift | |
|---|---|---|
| COOH | 14.3 ppm |  |
| NH | 13.8 ppm | 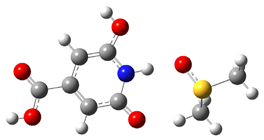 |
| OH | 13.5 ppm | 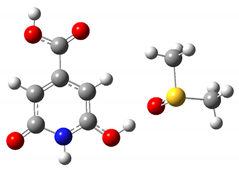 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mocci, F.; Olla, C.; Cappai, A.; Corpino, R.; Ricci, P.C.; Chiriu, D.; Salis, M.; Carbonaro, C.M. Formation of Citrazinic Acid Ions and Their Contribution to Optical and Magnetic Features of Carbon Nanodots: A Combined Experimental and Computational Approach. Materials 2021, 14, 770. https://doi.org/10.3390/ma14040770
Mocci F, Olla C, Cappai A, Corpino R, Ricci PC, Chiriu D, Salis M, Carbonaro CM. Formation of Citrazinic Acid Ions and Their Contribution to Optical and Magnetic Features of Carbon Nanodots: A Combined Experimental and Computational Approach. Materials. 2021; 14(4):770. https://doi.org/10.3390/ma14040770
Chicago/Turabian StyleMocci, Francesca, Chiara Olla, Antonio Cappai, Riccardo Corpino, Pier Carlo Ricci, Daniele Chiriu, Marcello Salis, and Carlo Maria Carbonaro. 2021. "Formation of Citrazinic Acid Ions and Their Contribution to Optical and Magnetic Features of Carbon Nanodots: A Combined Experimental and Computational Approach" Materials 14, no. 4: 770. https://doi.org/10.3390/ma14040770
APA StyleMocci, F., Olla, C., Cappai, A., Corpino, R., Ricci, P. C., Chiriu, D., Salis, M., & Carbonaro, C. M. (2021). Formation of Citrazinic Acid Ions and Their Contribution to Optical and Magnetic Features of Carbon Nanodots: A Combined Experimental and Computational Approach. Materials, 14(4), 770. https://doi.org/10.3390/ma14040770