
Establishing the Carbonation Profile with Raman Spectroscopy: Effects of Fly Ash and Ground Granulated Blast Furnace Slag



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Manufacture of FA/PC and GGBS/PC Blends
2.3. Curing, Conditioning and Carbonation
2.4. Bench-Mounted Raman Spectroscopy
2.5. Background Subtraction
2.6. Calculation of the Signal-to-Noise Ratio (SNR)
2.7. Thermogravimetry (TG)
3. Results and Discussion
- Establish the carbonation profiles viz. the variation of CaCO3 content against the depth of FA or GGBS containing PC pastes after being subjected to an accelerated carbonation with bench-mounted Raman spectroscopy.
- Verify the carbonation profiles as developed by Raman spectroscopy at stage 1 by the well-established TG analysis so that the potential effects that FA or GGBS may have on the reliability of the results obtained from Raman spectroscopy could be recognized.
3.1. Establishing the Carbonation Profiles of FA/PC and GGBS/PC Pastes with Bench-Mounted Raman Spectroscopy
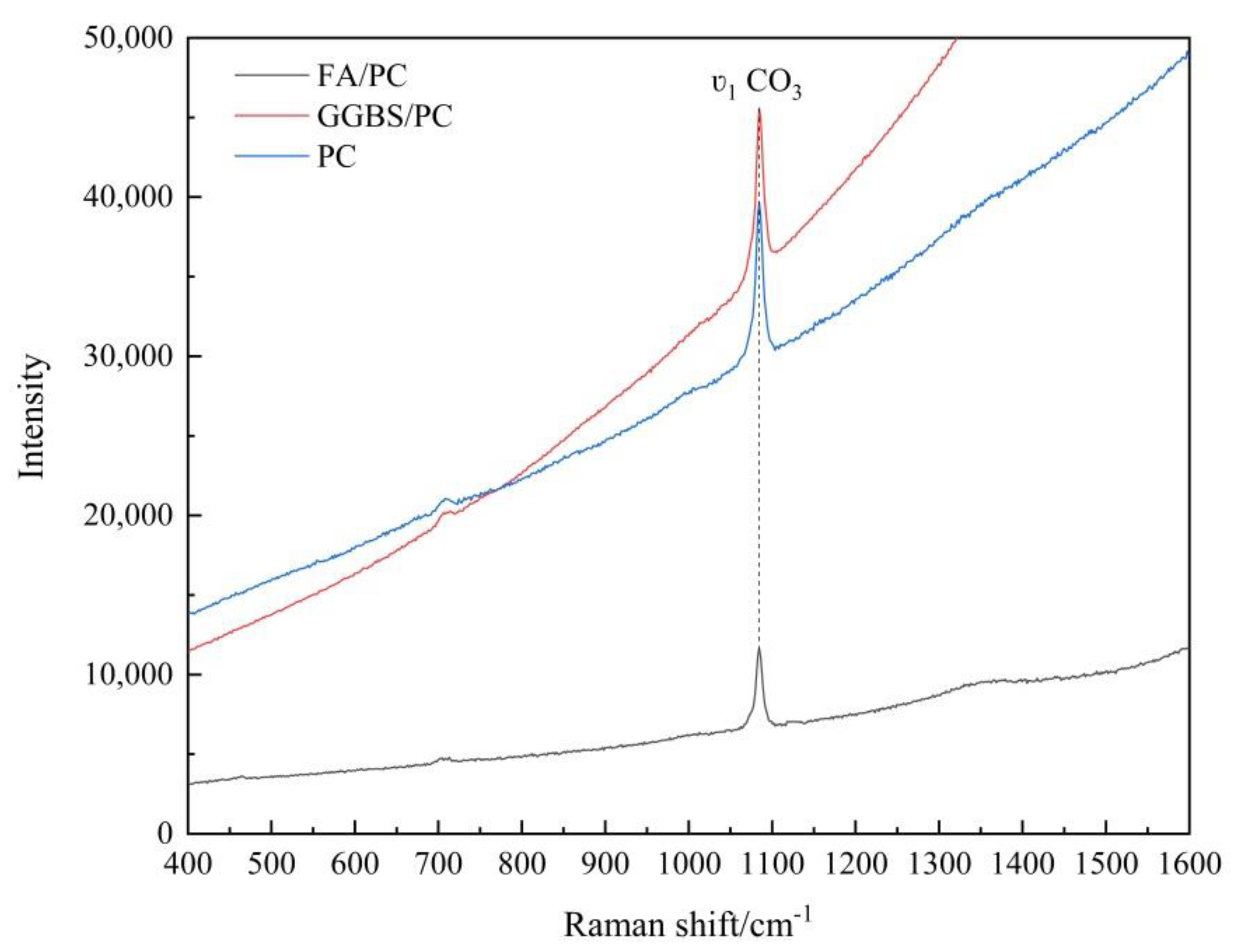
3.1.1. Raman Spectroscopy Analysis
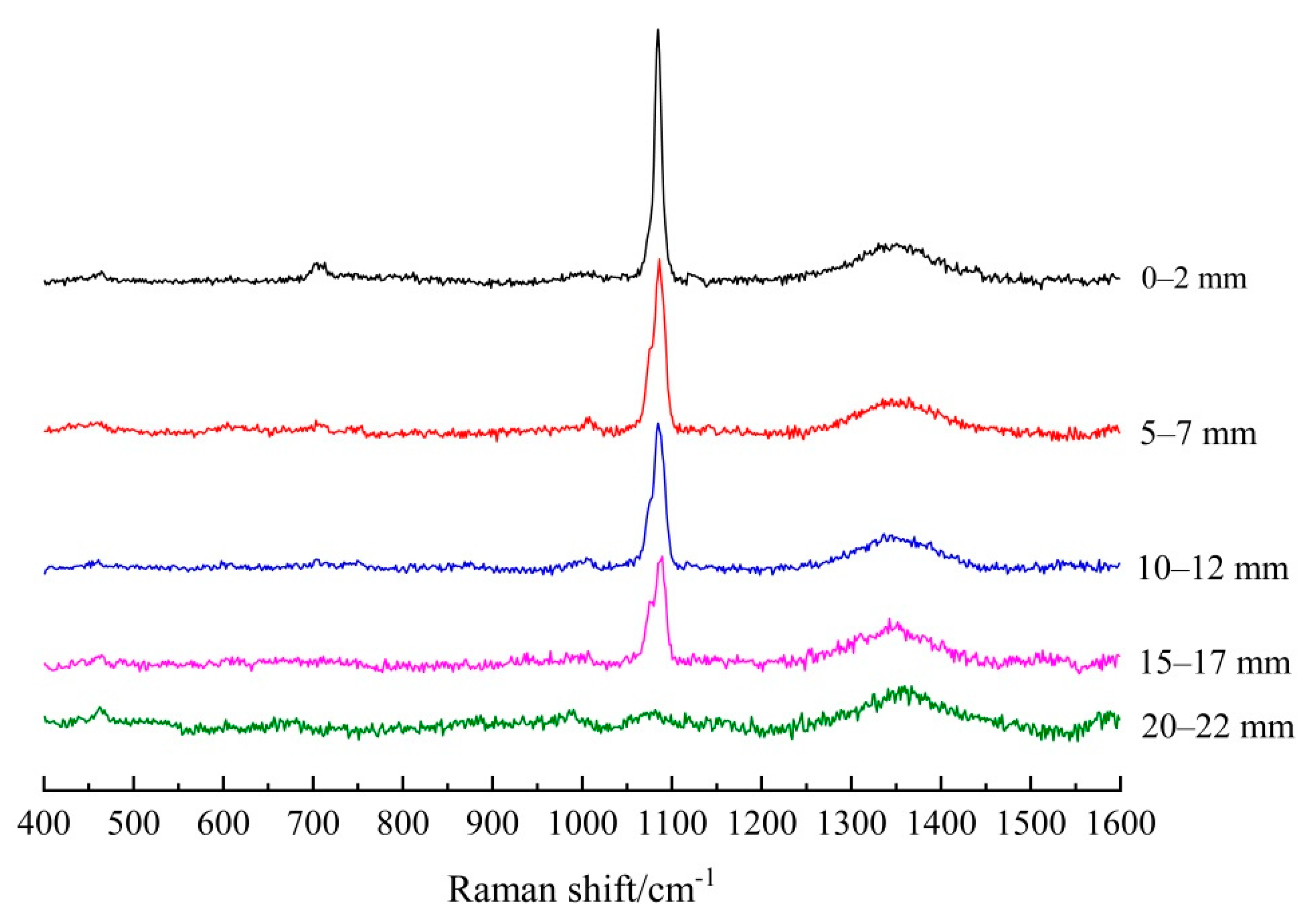
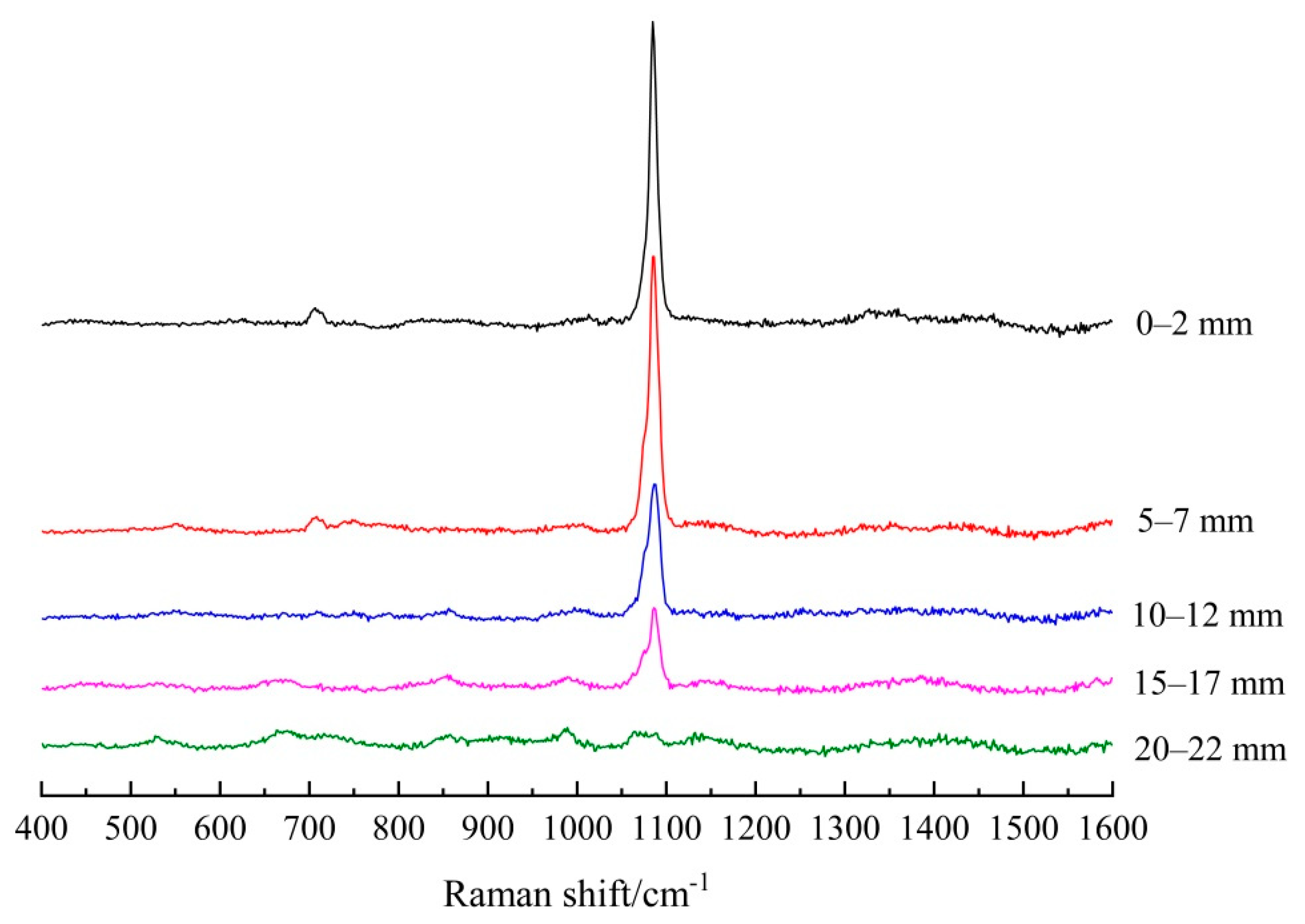
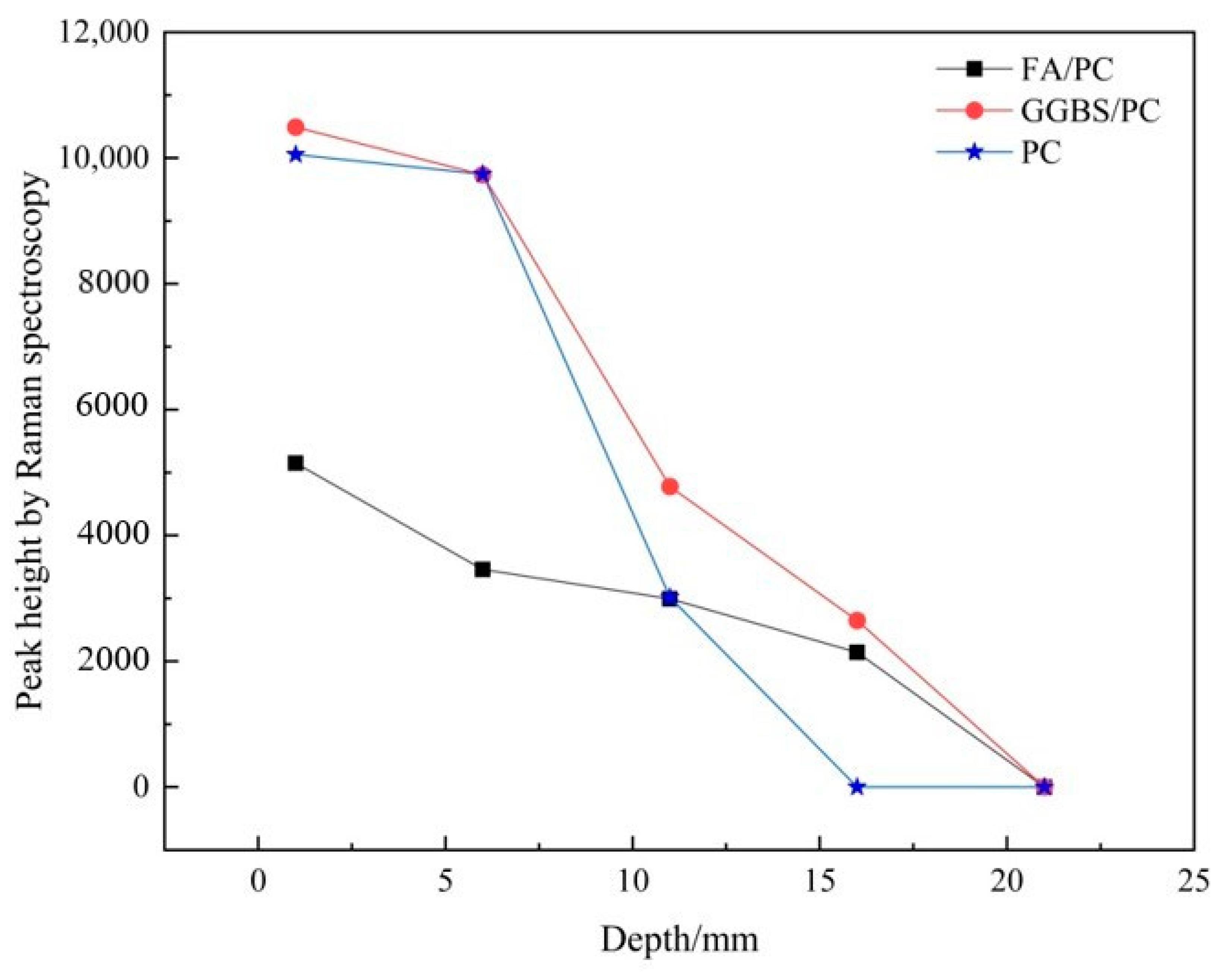
3.1.2. Establishing the Carbonation Profiles
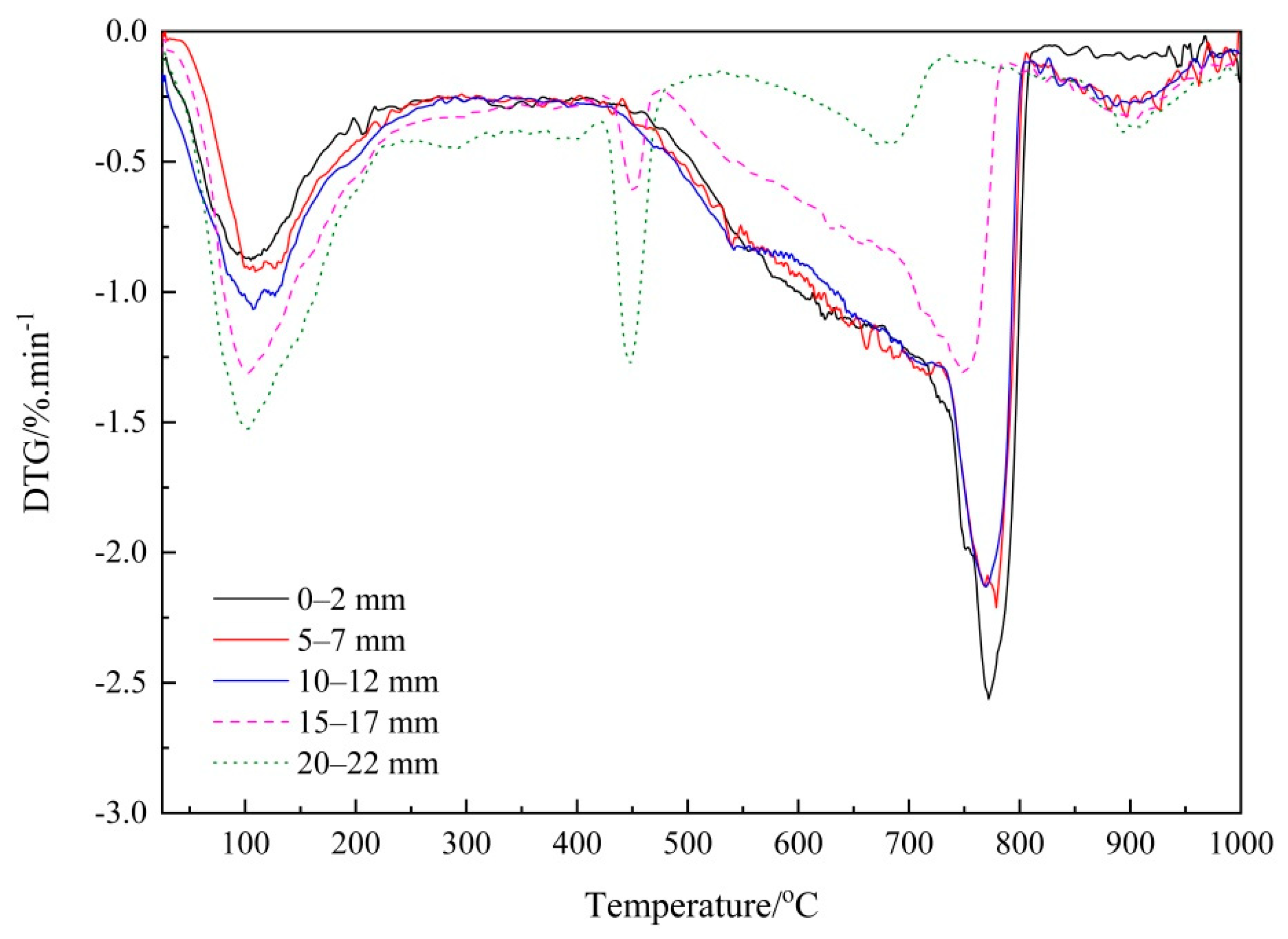
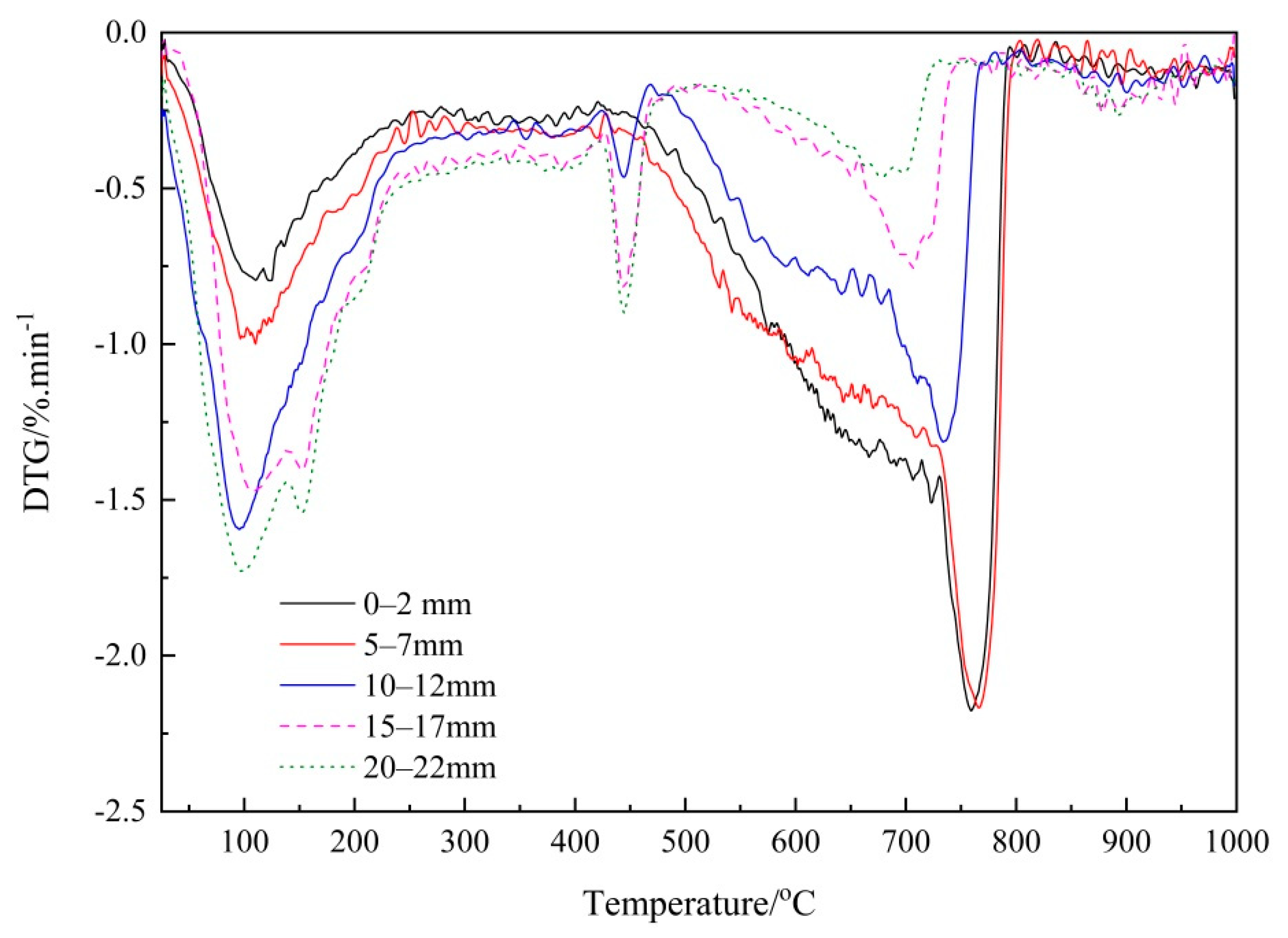
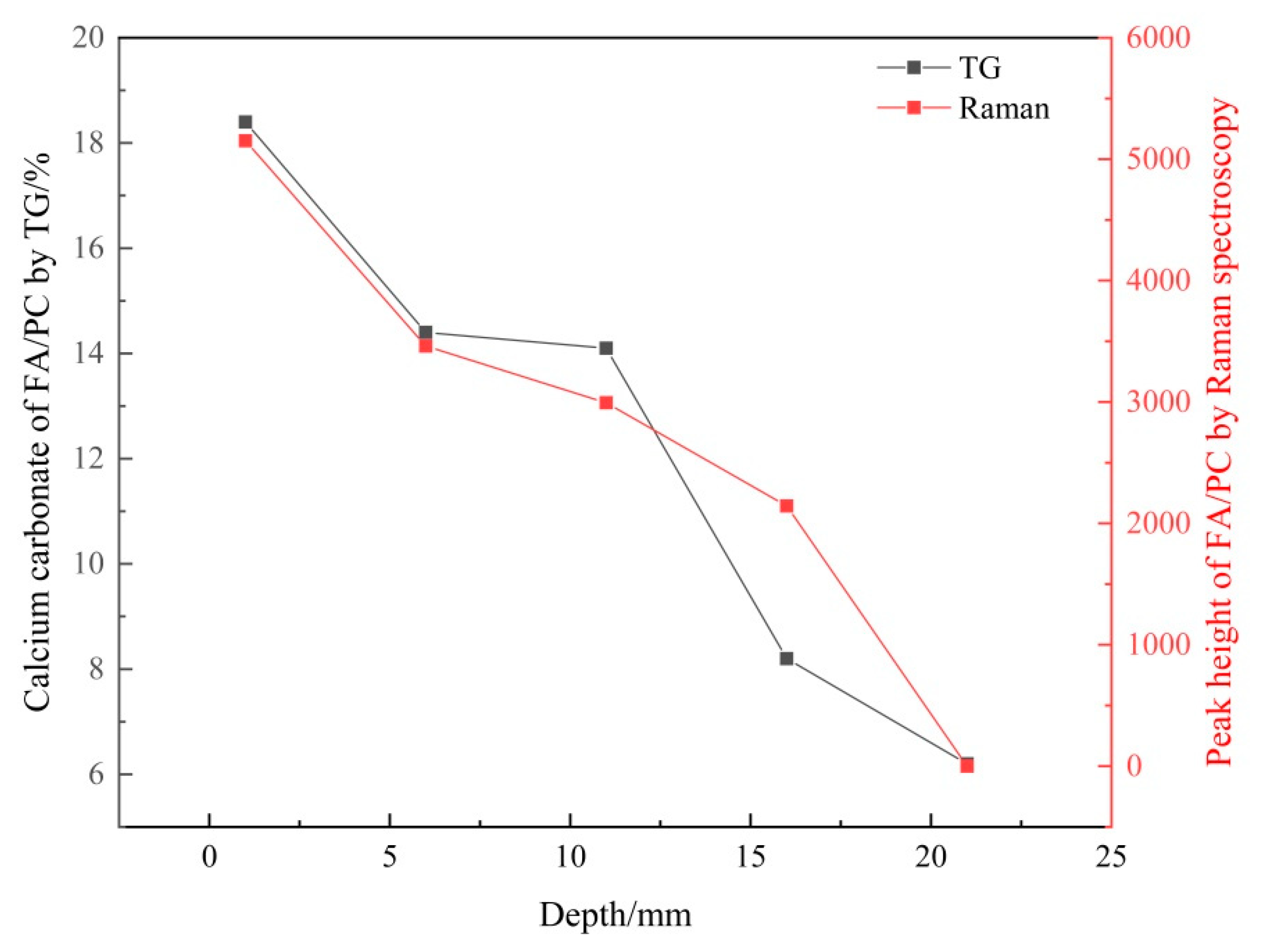
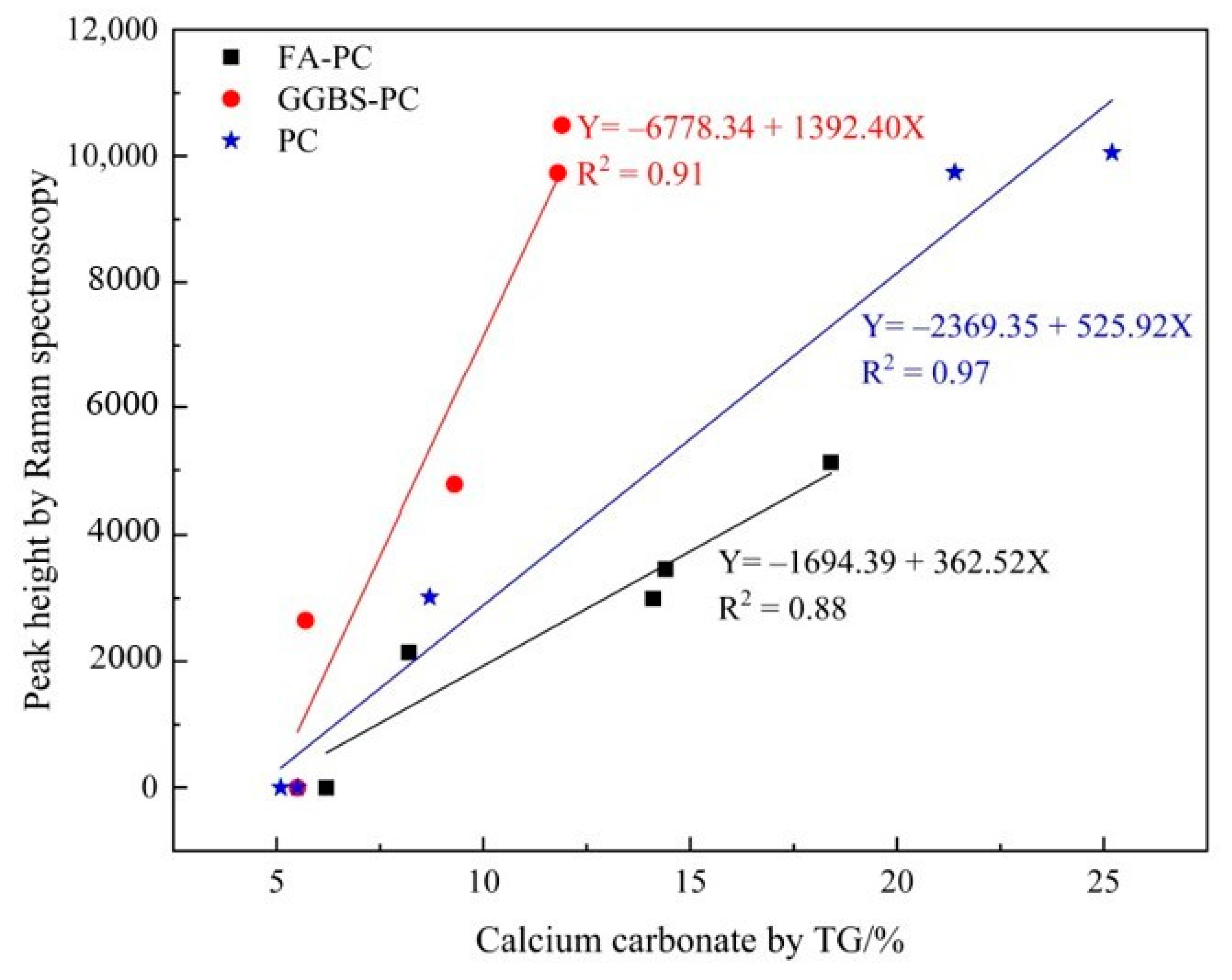
3.2. Verifying the Carbonation Profiles by TG Analysis
3.2.1. Thermogravimetry (TG) Analysis
3.2.2. Verifying the Carbonation Profiles
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neville, A.M. Properties of Concrete, 5th ed.; Prentice Hall: San Francisco, CA, USA, 1995. [Google Scholar]
- Šavija, B.; Luković, M. Carbonation of cement paste: Understanding, challenges, and opportunities. Construct. Build. Mater. 2016, 117, 285–301. [Google Scholar] [CrossRef]
- Broomfield, J.P. Carbonation and its effects in reinforced concrete. Mater. Perform. 2000, 39, 64–67. [Google Scholar]
- Sevelsted, T.F.; Skibsted, J. Carbonation of C–S–H and C–A–S–H samples studied by 13C, 27Al and 29Si MAS NMR spectroscopy. Cem. Concr. Res. 2015, 71, 56–65. [Google Scholar] [CrossRef]
- Villain, G.; Thiery, M.; Platret, G. Measurement methods of carbonation profiles in concrete: Thermogravimetry, chemical analysis and gammadensimetry. Cem. Concr. Res. 2007, 37, 1182–1192. [Google Scholar] [CrossRef]
- Morandeau, A.; Thiéry, M.; Dangla, P. Investigation of the carbonation mechanism of CH and C-S-H in terms of kinetics, microstructure changes and moisture properties. Cem. Concr. Res. 2014, 56, 153–170. [Google Scholar] [CrossRef]
- Liu, W.; Li, Y.Q.; Tang, L.P.; Dong, Z.J. XRD and 29Si MAS NMR study on carbonated cement paste under accelerated carbonation using different concentration of CO2. Mater. Today Commun. 2019, 19, 464–470. [Google Scholar] [CrossRef]
- Ruggieri, S.; Porco, F.; Uva, G.; Vamvatsikos, D. Two frugal options to assess class fragility and seismic safety for low-rise reinforced concrete school buildings in Southern Italy. Bull. Earthq. Eng. 2021, 19, 1415–1439. [Google Scholar] [CrossRef]
- Porco, F.; Ruggieri, S.; Uva, G. Seismic assessment of irregular existing building: Appraisal of the influence of compressive strength variation by means of nonlinear conventional and multimodal static analysis. Ing. Sismica 2018, 35. [Google Scholar]
- Houst, Y.F.; Wittmann, F.H. Depth profiles of carbonates formed during natural carbonation. Cem. Concr. Res. 2002, 32, 1923–1930. [Google Scholar] [CrossRef]
- Ann, K.Y.; Pack, S.W.; Hwang, J.P.; Song, H.W.; Kim, S.H. Service life prediction of a concrete bridge structure subjected to carbonation. Constr. Build. Mater. 2010, 24, 1494–1501. [Google Scholar] [CrossRef]
- Johannesson, B.; Utgenannt, P. Microstructural changes caused by carbonation of cement mortar. Cem. Concr. Res. 2001, 31, 925–931. [Google Scholar] [CrossRef]
- Taylor, H.F.W. Cement Chemistry, 2nd ed.; Thomas Telford: London, UK, 1997. [Google Scholar]
- Lee, H.J.; Kim, D.G.; Lee, J.H.; Cho, M.S. A study for carbonation degree on concrete using a phenolphthalein indicator and fourier-transform infrared spectroscopy. World Acad. Sci. Eng. Technol. 2012, 62, 184–190. [Google Scholar]
- Lo, Y.; Lee, H.M. Curing effects on carbonation of concrete using a phenolphthalein indicator and Fourier-transform infrared spectroscopy. Build. Environ. 2002, 37, 507–514. [Google Scholar] [CrossRef]
- Thiery, M.; Villain, G.; Dangla, P.; Platret, G. Investigation of the carbonation front shape on cementitious materials: Effects of the chemical kinetics. Cem. Concr. Res. 2007, 37, 1047–1058. [Google Scholar] [CrossRef]
- Chang, C.-F.; Chen, J.-W. The experimental investigation of concrete carbonation depth. Cem. Concr. Res. 2006, 36, 1760–1767. [Google Scholar] [CrossRef]
- Long, D.A. Raman Spectroscopy; McGraw-Hill International Book Company: New York, NY, USA, 1977. [Google Scholar]
- McCreery, R.L. Raman Spectroscopy for Chemical Analysis; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2000. [Google Scholar]
- Predeanu, G.; Popescu, L.G.; Abagiu, T.A.; Panaitescu, C.; Valentim, B.; Guedes, A. Characterization of bottom ash of Pliocene lignite as ceramic composites raw material by petrographic, SEM/EDS and Raman microspectroscopical methods. Int. J. Coal Geol. 2016, 168, 131–145. [Google Scholar] [CrossRef]
- Corvisier, J.; Brunet, F.; Fabbri, A.; Bernard, S.; Findling, N.; Rimmelé, G.; Barlet-Gouédard, V.; Beyssac, O.; Goffé, B. Raman mapping and numerical simulation of calcium carbonates distribution in experimentally carbonated Portland-cement cores. Eur. J. Miner. 2010, 22, 63–74. [Google Scholar] [CrossRef]
- Yue, Y.; Wang, J.J.; Basheer, P.A.M.; Bai, Y. Raman spectroscopic investigation of Friedel’s salt. Cem. Concr. Compos. 2018, 86, 306–314. [Google Scholar] [CrossRef]
- Yue, Y.; Wang, J.J.; Basheer, P.A.M.; Boland, J.J.; Bai, Y. Characterisation of carbonated Portland cement paste with optical fibre excitation Raman spectroscopy. Constr. Build. Mater. 2017, 135, 369–376. [Google Scholar] [CrossRef]
- Yue, Y.; Wang, J.J.; Bai, Y. Tracing the status of silica fume in cementitious materials with Raman microscope. Constr. Build. Mater. 2018, 159, 610–616. [Google Scholar] [CrossRef]
- Yue, Y.; Bai, Y.; Basheer, P.A.M.; Boland, J.J.; Wang, J.J. Monitoring the cementitious materials subjected to sulfate attack with optical fiber excitation Raman spectroscopy. Opt. Eng. 2013, 52, 104107. [Google Scholar] [CrossRef]
- Yue, Y.; Wang, J.J.; Basheer, P.A.M.; Boland, J.J.; Bai, Y. A Raman spectroscopy based optical fibre system for detecting carbonation profile of cementitious materials. Sens. Actuator. B Chem. 2018, 257, 635–649. [Google Scholar] [CrossRef]
- Potgieter-Vermaak, S.S.; Potgieter, J.H.; Belleil, M.; DeWeerdt, F.; Van Grieken, R. The application of Raman spectrometry to the investigation of cement: Part II: A micro-Raman study of OPC, slag and fly ash. Cem. Concr. Res. 2006, 36, 663–670. [Google Scholar] [CrossRef]
- Martinez-Ramirez, S.; Sanchez-Cortes, S.; Garcia-Ramos, J.V.; Domingo, C.; Fortes, C.; Blanco-Varela, M.T. Micro-Raman spectroscopy applied to depth profiles of carbonates formed in lime mortar. Cem. Concr. Res. 2003, 33, 2063–2068. [Google Scholar] [CrossRef]
- Black, L.; Breen, C.; Yarwood, J.; Garbev, K.; Gasharova, B. Structural Features of C–S–H(I) and Its Carbonation in Air—A Raman Spectroscopic Study. Part II: Carbonated Phases. J. Am. Ceram. Soc. 2007, 90, 908–917. [Google Scholar] [CrossRef]
- Tlili, M.M.; Ben Amor, M.; Gabrielli, C.; Joiret, S.; Maurin, G.; Rousseau, P. Characterization of CaCO3 hydrates by micro-Raman spectroscopy. J. Raman Spectrosc. 2002, 33, 10–16. [Google Scholar] [CrossRef]
- Ibanez, J.; Artus, L.; Cusco, R.; Lopez, A.; Menendez, E.; Andrade, M.C. Hydration and carbonation of monoclinic C2S and C3S studied by Raman spectroscopy. J. Raman Spectrosc. 2007, 38, 61–67. [Google Scholar] [CrossRef]
- Caggiani, M.C.; Colomban, P. Raman identification of strongly absorbing phases: The ceramic black pigments. J. Raman Spectrosc. 2011, 42, 839–843. [Google Scholar] [CrossRef]
- Richardson, I.G.; Skibsted, J.; Black, L.; Kirkpatrick, R.J. Characterisation of cement hydrate phases by TEM, NMR and Raman spectroscopy. Adv. Cem. Res. 2010, 22, 233–248. [Google Scholar] [CrossRef]
- Hussain, S.; Bhunia, D.; Singh, S.B. Comparative study of accelerated carbonation of plain cement and fly-ash concrete. J. Build. Eng. 2017, 10, 26–31. [Google Scholar] [CrossRef]
- Han-Seung, L.; Wang, X.-Y. Evaluation of compressive strength development and carbonation depth of high volume slag-blended concrete. Constr. Build. Mater. 2016, 124, 45–54. [Google Scholar] [CrossRef]
- Sun, Q.; Qin, C. Raman OH stretching band of water as an internal standard to determine carbonate concentrations. Chem. Geol. 2011, 283, 274–278. [Google Scholar] [CrossRef]
- Moreno, T.; Morán López, M.A.; Huerta Illera, I.; Piqueras, C.M.; Sanz Arranz, A.; García Serna, J.; Cocero, M.J. Quantitative Raman determination of hydrogen peroxide using the solvent as internal standard: Online application in the direct synthesis of hydrogen peroxide. Chem. Eng. J. 2011, 166, 1061–1065. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxides/% | SiO2 | Al2O3 | Fe2O3 | CaO | MgO | K2O | Na2O | SO3 |
|---|---|---|---|---|---|---|---|---|
| PC | 23.00 | 6.15 | 2.95 | 61.30 | 1.80 | 0.68 | 0.22 | 2.50 |
| FA | 56.30 | 23.50 | 4.70 | 4.40 | 1.80 | 1.80 | 1.00 | 0.90 |
| GGBS | 34.30 | 15.00 | 0.40 | 39.40 | 8.00 | 0.38 | 0.45 | 0.80 |
| Parameters of Spectra | FA/PC Paste | GGBS/PC Paste | PC Paste |
|---|---|---|---|
| Signal (peak intensity) | 5151.5 (51%) | 10,491.3 (104%) | 10,055.1 (100%) |
| Noise | 110.0 (89%) | 139.8 (113%) | 124.2 (100%) |
| SNR | 47 | 75 | 81 |
| Depth/mm | FA/PC Paste | GGBS/PC Paste | PC Paste |
|---|---|---|---|
| 0–2 | 5151.5 | 10491.3 | 10,055.1 |
| 5–7 | 3461.5 | 9734.5 | 9741.7 |
| 10–12 | 2993.9 | 4776.6 | 3014.8 |
| 15–17 | 2143.7 | 2650.0 | 0 |
| 20–22 | 0 | 0 | 0 |
| Depth/mm | FA/PC Paste | GGBS/PC Paste | PC Paste |
|---|---|---|---|
| 0–2 | 18.4 | 11.9 | 25.2 |
| 5–7 | 14.4 | 11.8 | 21.4 |
| 10–12 | 14.1 | 9.3 | 8.7 |
| 15–17 | 8.2 | 5.7 | 5.5 |
| 20–22 | 6.2 | 5.5 | 5.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yue, Y.; Wang, J.J.; Basheer, P.A.M.; Bai, Y. Establishing the Carbonation Profile with Raman Spectroscopy: Effects of Fly Ash and Ground Granulated Blast Furnace Slag. Materials 2021, 14, 1798. https://doi.org/10.3390/ma14071798
Yue Y, Wang JJ, Basheer PAM, Bai Y. Establishing the Carbonation Profile with Raman Spectroscopy: Effects of Fly Ash and Ground Granulated Blast Furnace Slag. Materials. 2021; 14(7):1798. https://doi.org/10.3390/ma14071798
Chicago/Turabian StyleYue, Yanfei, Jing Jing Wang, P. A. Muhammed Basheer, and Yun Bai. 2021. "Establishing the Carbonation Profile with Raman Spectroscopy: Effects of Fly Ash and Ground Granulated Blast Furnace Slag" Materials 14, no. 7: 1798. https://doi.org/10.3390/ma14071798
APA StyleYue, Y., Wang, J. J., Basheer, P. A. M., & Bai, Y. (2021). Establishing the Carbonation Profile with Raman Spectroscopy: Effects of Fly Ash and Ground Granulated Blast Furnace Slag. Materials, 14(7), 1798. https://doi.org/10.3390/ma14071798