
Crumpled Graphene-Storage Media for Hydrogen and Metal Nanoclusters

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Graphene with Nickel Nanocluster
2.2. Graphene with Hydrogen Nanocluster
3. Results
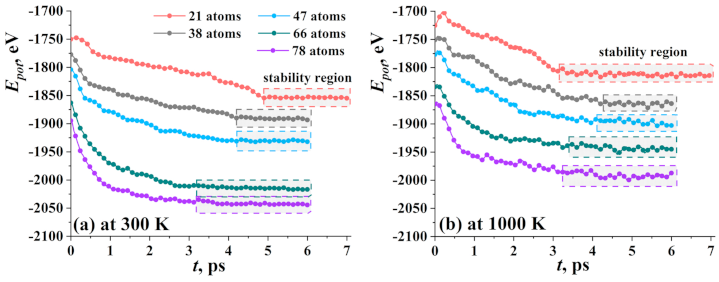
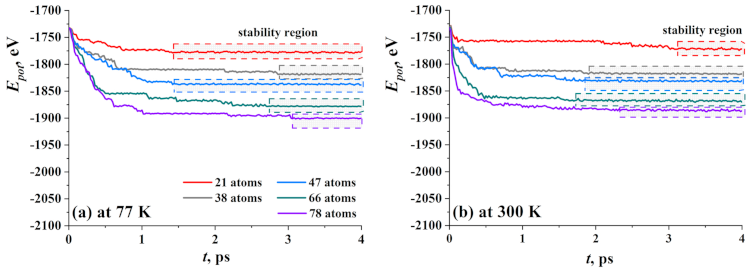
3.1. Graphene with Ni Nanocluster
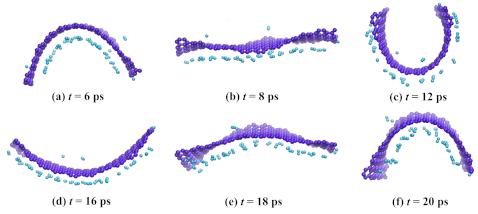
3.2. Graphene with Hydrogen Nanocluster
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MD | Molecular Dynamics |
| CNT | Carbon Nanotubes |
| GF | Graphene Flake |
| SSA | Specific Surface Area |
References
- Iwan, A.; Malinowski, M.; Pasciak, G. Polymer fuel cell components modified by graphene: Electrodes, electrolytes and bipolar plates. Renew. Sustain. Energy Rev. 2015, 49, 954–967. [Google Scholar] [CrossRef]
- Baimova, J.A.; Rysaeva, L.K.; Liu, B.; Dmitriev, S.V.; Zhou, K. From flat graphene to bulk carbon nanostructures. Phys. Status Solidi (b) 2015, 252, 1502–1507. [Google Scholar] [CrossRef]
- Xiao, J.; Mei, D.; Li, X.; Xu, W.; Wang, D.; Graff, G.L.; Bennett, W.D.; Nie, Z.; Saraf, L.V.; Aksay, I.A.; et al. Hierarchically Porous Graphene as a Lithium–Air Battery Electrode. Nano Lett. 2011, 11, 5071–5078. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Cheng, B.; Hu, X.; Yu, J. Holey Graphene for Electrochemical Energy Storage. Cell Rep. Phys. Sci. 2020, 1, 100215. [Google Scholar] [CrossRef]
- Baimova, Y.A.; Murzaev, R.T.; Dmitriev, S.V. Mechanical properties of bulk carbon nanomaterials. Phys. Solid State 2014, 56, 2010–2016. [Google Scholar] [CrossRef]
- Niu, L.; Xie, J.; Chen, P.; Li, G.; Zhang, X. Quasi-static compression properties of graphene aerogel. Diam. Relat. Mater. 2021, 111, 108225. [Google Scholar] [CrossRef]
- Yola, M.L. Development of Novel Nanocomposites Based on Graphene/Graphene Oxide and Electrochemical Sensor Applications. Curr. Anal. Chem. 2019, 15, 159–165. [Google Scholar] [CrossRef]
- Meyer, J.C.; Geim, A.K.; Katsnelson, M.I.; Novoselov, K.S.; Booth, T.J.; Roth, S. The structure of suspended graphene sheets. Nature 2007, 446, 60–63. [Google Scholar] [CrossRef]
- Tallinen, T.; Åström, J.A.; Timonen, J. The effect of plasticity in crumpling of thin sheets. Nat. Mater. 2008, 8, 25–29. [Google Scholar] [CrossRef]
- Matan, K.; Williams, R.B.; Witten, T.A.; Nagel, S.R. Crumpling a Thin Sheet. Phys. Rev. Lett. 2002, 88. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, F.; Yang, X.; Long, G.; Wu, Y.; Zhang, T.; Leng, K.; Huang, Y.; Ma, Y.; Yu, A.; et al. Porous 3D graphene-based bulk materials with exceptional high surface area and excellent conductivity for supercapacitors. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [Green Version]
- Balankin, A.S.; Ochoa, D.S.; León, E.P.; de Oca, R.C.M.; Rangel, A.H.; Cruz, M.Á.M. Power law scaling of lateral deformations with universal Poisson’s index for randomly folded thin sheets. Phys. Rev. B 2008, 77. [Google Scholar] [CrossRef]
- Baimova, J.A.; Liu, B.; Zhou, K. Folding and crumpling of graphene under biaxial compression. Lett. Mater. 2014, 4, 96–99. [Google Scholar] [CrossRef] [Green Version]
- Krainyukova, N.V.; Zubarev, E.N. Carbon Honeycomb High Capacity Storage for Gaseous and Liquid Species. Phys. Rev. Lett. 2016, 116. [Google Scholar] [CrossRef]
- Semat, J.M.; Rashmi, W.; Mahesh, V.; Khalid, M.; Priyanka, J. Synthesis of crumpled graphene by fast cooling method. In Proceedings of the International Engineering Research Conference-12th Eureca 2019; AIP Publishing: Melville, NY, USA, 2019. [Google Scholar] [CrossRef]
- Savin, A.; Savina, O.I. The Effect of Layers Interaction on the Stiffness of Bending Deformations of Multilayered Carbon Nanoribbons. Phys. Solid State 2019, 61, 686–692. [Google Scholar] [CrossRef]
- van Bruggen, E.; van der Linden, E.; Habibi, M. Tailoring relaxation dynamics and mechanical memory of crumpled materials by friction and ductility. Soft Matter 2019, 15, 1633–1639. [Google Scholar] [CrossRef]
- Liao, Y.; Li, Z.; Xia, W. Size-dependent structural behaviors of crumpled graphene sheets. Carbon 2021, 174, 148–157. [Google Scholar] [CrossRef]
- Liu, S.; Wang, A.; Li, Q.; Wu, J.; Chiou, K.; Huang, J.; Luo, J. Crumpled Graphene Balls Stabilized Dendrite-free Lithium Metal Anodes. Joule 2018, 2, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Jang, H.D.; Sun, T.; Xiao, L.; He, Z.; Katsoulidis, A.P.; Kanatzidis, M.G.; Gibson, J.M.; Huang, J. Compression and Aggregation-Resistant Particles of Crumpled Soft Sheets. ACS Nano 2011, 5, 8943–8949. [Google Scholar] [CrossRef]
- Baimova, J.A.; Liu, B.; Dmitriev, S.V.; Zhou, K. Mechanical properties of crumpled graphene under hydrostatic and uniaxial compression. J. Phys. D Appl. Phys. 2015, 48, 095302. [Google Scholar] [CrossRef]
- Khan, M.B.; Wang, C.; Wang, S.; Fang, D.; Chen, S. The mechanical property and microscopic deformation mechanism of nanoparticle-contained graphene foam materials under uniaxial compression. Nanotechnology 2020, 32, 115701. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Ren, P.; Deng, D.; Deng, J.; Bao, X. Single layer graphene encapsulating non-precious metals as high-performance electrocatalysts for water oxidation. Energy Environ. Sci. 2016, 9, 123–129. [Google Scholar] [CrossRef]
- Wang, X.X.; Tan, Z.H.; Zeng, M.; Wang, J.N. Carbon nanocages: A new support material for Pt catalyst with remarkably high durability. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Ren, P.; Deng, D.; Bao, X. Enhanced Electron Penetration through an Ultrathin Graphene Layer for Highly Efficient Catalysis of the Hydrogen Evolution Reaction. Angew. Chem. Int. Ed. 2015, 54, 2100–2104. [Google Scholar] [CrossRef] [PubMed]
- Dahal, A.; Batzill, M. Graphene–nickel interfaces: A review. Nanoscale 2014, 6, 2548. [Google Scholar] [CrossRef] [PubMed]
- Sutter, P.; Sadowski, J.T.; Sutter, E. Graphene on Pt(111): Growth and substrate interaction. Phys. Rev. B 2009, 80, 245411. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Guest, J.R.; Guisinger, N.P. Epitaxial Graphene on Cu(111). Nano Lett. 2010, 10, 3512–3516. [Google Scholar] [CrossRef]
- Yoon, S.M.; Choi, W.M.; Baik, H.; Shin, H.J.; Song, I.; Kwon, M.S.; Bae, J.J.; Kim, H.; Lee, Y.H.; Choi, J.Y. Synthesis of Multilayer Graphene Balls by Carbon Segregation from Nickel Nanoparticles. ACS Nano 2012, 6, 6803–6811. [Google Scholar] [CrossRef]
- Ji, Z.; Wang, Y.; Yu, Q.; Shen, X.; Li, N.; Ma, H.; Yang, J.; Wang, J. One-step thermal synthesis of nickel nanoparticles modified graphene sheets for enzymeless glucose detection. J. Colloid Interface Sci. 2017, 506, 678–684. [Google Scholar] [CrossRef]
- Ai, L.; Tian, T.; Jiang, J. Ultrathin Graphene Layers Encapsulating Nickel Nanoparticles Derived Metal–Organic Frameworks for Highly Efficient Electrocatalytic Hydrogen and Oxygen Evolution Reactions. ACS Sustain. Chem. Eng. 2017, 5, 4771–4777. [Google Scholar] [CrossRef]
- Batzill, M. The surface science of graphene: Metal interfaces, CVD synthesis, nanoribbons, chemical modifications, and defects. Surf. Sci. Rep. 2012, 67, 83–115. [Google Scholar] [CrossRef]
- Omukai, A.; Yoshimura, A.; Watanabe, F.; Tachibana, M. Graphene films synthesized by chemical vapor deposition with ethanol. Trans. Mater. Res. Soc. Jpn. 2011, 36, 359–362. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Cai, W.; Colombo, L.; Ruoff, R.S. Evolution of Graphene Growth on Ni and Cu by Carbon Isotope Labeling. Nano Lett. 2009, 9, 4268–4272. [Google Scholar] [CrossRef] [Green Version]
- Chae, S.J.; Güneş, F.; Kim, K.K.; Kim, E.S.; Han, G.H.; Kim, S.M.; Shin, H.J.; Yoon, S.M.; Choi, J.Y.; Park, M.H.; et al. Synthesis of Large-Area Graphene Layers on Poly-Nickel Substrate by Chemical Vapor Deposition: Wrinkle Formation. Adv. Mater. 2009, 21, 2328–2333. [Google Scholar] [CrossRef]
- Reina, A.; Thiele, S.; Jia, X.; Bhaviripudi, S.; Dresselhaus, M.S.; Schaefer, J.A.; Kong, J. Growth of large-area single- and Bi-layer graphene by controlled carbon precipitation on polycrystalline Ni surfaces. Nano Res. 2009, 2, 509–516. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.W.; Nair, A.K.; Buehler, M.J. Nanoindentation study of size effects in nickel–graphene nanocomposites. Philos. Mag. Lett. 2013, 93, 196–203. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, J.; Yeom, M.S.; Shin, J.W.; Kim, H.; Cui, Y.; Kysar, J.W.; Hone, J.; Jung, Y.; Jeon, S.; et al. Strengthening effect of single-atomic-layer graphene in metal–graphene nanolayered composites. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Kuang, D.; Xu, L.; Liu, L.; Hu, W.; Wu, Y. Graphene–nickel composites. Appl. Surf. Sci. 2013, 273, 484–490. [Google Scholar] [CrossRef]
- Kim, D.J.; Truong, Q.T.; Kim, J.I.; Suh, Y.; Moon, J.; Lee, S.E.; Hong, B.H.; Woo, Y.S. Ultrahigh-strength multi-layer graphene-coated Ni film with interface-induced hardening. Carbon 2021, 178, 497–505. [Google Scholar] [CrossRef]
- Safina, L.R.; Baimova, J.A.; Mulyukov, R.R. Nickel nanoparticles inside carbon nanostructures: Atomistic simulation. Mech. Adv. Mater. Mod. Process. 2019, 5. [Google Scholar] [CrossRef]
- Safina, L.R.; Krylova, K.A. Effect of particle size on the formation of the composite structure in Ni-graphene system: Atomistic simulation. J. Phys. Conf. Ser. 2020, 1435, 012067. [Google Scholar] [CrossRef]
- Safina, L.L.; Baimova, J.A. Molecular dynamics simulation of fabrication of Ni-graphene composite: Temperature effect. Micro Nano Lett. 2020, 15, 176–180. [Google Scholar] [CrossRef]
- Safina, L.; Baimova, J.; Krylova, K.; Murzaev, R.; Mulyukov, R. Simulation of metal-graphene composites by molecular dynamics: A review. Lett. Mater. 2020, 10, 351–360. [Google Scholar] [CrossRef]
- Wu, C.D.; Fang, T.H.; Lo, J.Y. Effects of pressure, temperature, and geometric structure of pillared graphene on hydrogen storage capacity. Int. J. Hydrog. Energy 2012, 37, 14211–14216. [Google Scholar] [CrossRef]
- Krylova, K.; Baimova, J.; Mulyukov, R. Effect of deformation on dehydrogenation mechanisms of crumpled graphene: Molecular dynamics simulation. Lett. Mater. 2019, 9, 81–85. [Google Scholar] [CrossRef]
- Krylova, K.A.; Baimova, J.A.; Lobzenko, I.P.; Rudskoy, A.I. Crumpled graphene as a hydrogen storage media: Atomistic simulation. Phys. B Condens. Matter 2020, 583, 412020. [Google Scholar] [CrossRef]
- Dimitrakakis, G.K.; Tylianakis, E.; Froudakis, G.E. Pillared Graphene: A New 3-D Network Nanostructure for Enhanced Hydrogen Storage. Nano Lett. 2008, 8, 3166–3170. [Google Scholar] [CrossRef] [PubMed]
- Ranjbar, A.; Khazaei, M.; Venkataramanan, N.S.; Lee, H.; Kawazoe, Y. Chemical engineering of adamantane by lithium functionalization: A first-principles density functional theory study. Phys. Rev. B 2011, 83. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.T. Hydrogen storage by alkali-doped carbon nanotubes–revisited. Carbon 2000, 38, 623–626. [Google Scholar] [CrossRef]
- Khantha, M.; Cordero, N.A.; Molina, L.M.; Alonso, J.A.; Girifalco, L.A. Interaction of lithium with graphene: Anab initiostudy. Phys. Rev. B 2004, 70. [Google Scholar] [CrossRef]
- Medeiros, P.V.C.; de Brito Mota, F.; Mascarenhas, A.J.S.; de Castilho, C.M.C. Adsorption of monovalent metal atoms on graphene: A theoretical approach. Nanotechnology 2010, 21, 115701. [Google Scholar] [CrossRef]
- Yoshida, A.; Okuyama, T.; Terada, T.; Naito, S. Reversible hydrogen storage/release phenomena on lithium fulleride (LinC60) and their mechanistic investigation by solid-state NMR spectroscopy. J. Mater. Chem. 2011, 21, 9480. [Google Scholar] [CrossRef]
- Waqar, Z. Hydrogen accumulation in graphite and etching of graphite on hydrogen desorption. J. Mater. Chem. 2007, 42, 1169–1176. [Google Scholar] [CrossRef]
- oś, S.; Duclaux, L.; Letellier, M.; Azaïs, P. Study of Adsorption Properties on Lithium Doped Activated Carbon Materials. Acta Phys. Pol. A 2005, 108, 371–377. [Google Scholar] [CrossRef]
- Stuart, S.J.; Tutein, A.B.; Harrison, J.A. A reactive potential for hydrocarbons with intermolecular interactions. J. Chem. Phys. 2000, 112, 6472–6486. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Gragh. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Girifalco, L.A.; Weizer, V.G. Application of the Morse Potential Function to Cubic Metals. Phys. Rev. 1959, 114, 687–690. [Google Scholar] [CrossRef]
- Katin, K.P.; Prudkovskiy, V.S.; Maslov, M.M. Molecular dynamics simulation of nickel-coated graphene bending. Micro Nano Lett. 2018, 13, 160–164. [Google Scholar] [CrossRef]
- Galashev, A.Y.; Katin, K.P.; Maslov, M.M. Morse parameters for the interaction of metals with graphene and silicene. Phys. Lett. A 2019, 383, 252–258. [Google Scholar] [CrossRef]
- Petucci, J.; LeBlond, C.; Karimi, M.; Vidali, G. Diffusion, adsorption, and desorption of molecular hydrogen on graphene and in graphite. J. Chem. Phys. 2013, 139, 044706. [Google Scholar] [CrossRef] [Green Version]
- Ganz, E.; Ganz, A.B.; Yang, L.M.; Dornfeld, M. The initial stages of melting of graphene between 4000 K and 6000 K. Phys. Chem. Chem. Phys. 2017, 19, 3756–3762. [Google Scholar] [CrossRef] [PubMed]
- Los, J.H.; Zakharchenko, K.V.; Katsnelson, M.I.; Fasolino, A. Melting temperature of graphene. Phys. Rev. B 2015, 91, 045415. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Ahn, C.C.; Witham, C.; Fultz, B.; Liu, J.; Rinzler, A.G.; Colbert, D.; Smith, K.A.; Smalley, R.E. Hydrogen adsorption and cohesive energy of single-walled carbon nanotubes. Appl. Phys. Lett. 1999, 74, 2307–2309. [Google Scholar] [CrossRef] [Green Version]
- Pinkerton, F.E.; Wicke, B.G.; Olk, C.H.; Tibbetts, G.G.; Meisner, G.P.; Meyer, M.S.; Herbst, J.F. Thermogravimetric Measurement of Hydrogen Absorption in Alkali-Modified Carbon Materials. J. Phys. Chem. B 2000, 104, 9460–9467. [Google Scholar] [CrossRef]
- Schimmel, H.G.; Kearley, G.J.; Nijkamp, M.G.; Visser, C.T.; de Jong, K.P.; Mulder, F.M. Hydrogen Adsorption in Carbon Nanostructures: Comparison of Nanotubes, Fibers, and Coals. Chem. A Eur. J. 2003, 9, 4764–4770. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D, Å | N | d, Å | a, Å |
|---|---|---|---|
| 21 | 6.2 | 4.25 | |
| 38 | 8.1 | 3.30 | |
| 14.7 | 47 | 9.0 | 2.85 |
| 66 | 9.4 | 2.65 | |
| 78 | 10.4 | 2.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Safina, L.R.; Krylova, K.A.; Murzaev, R.T.; Baimova, J.A.; Mulyukov, R.R. Crumpled Graphene-Storage Media for Hydrogen and Metal Nanoclusters. Materials 2021, 14, 2098. https://doi.org/10.3390/ma14092098
Safina LR, Krylova KA, Murzaev RT, Baimova JA, Mulyukov RR. Crumpled Graphene-Storage Media for Hydrogen and Metal Nanoclusters. Materials. 2021; 14(9):2098. https://doi.org/10.3390/ma14092098
Chicago/Turabian StyleSafina, Liliya R., Karina A. Krylova, Ramil T. Murzaev, Julia A. Baimova, and Radik R. Mulyukov. 2021. "Crumpled Graphene-Storage Media for Hydrogen and Metal Nanoclusters" Materials 14, no. 9: 2098. https://doi.org/10.3390/ma14092098