
The Role of Zr on Monoclinic and Orthorhombic HfxZryO2 Systems: A First-Principles Study

 ,
, 
 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussions
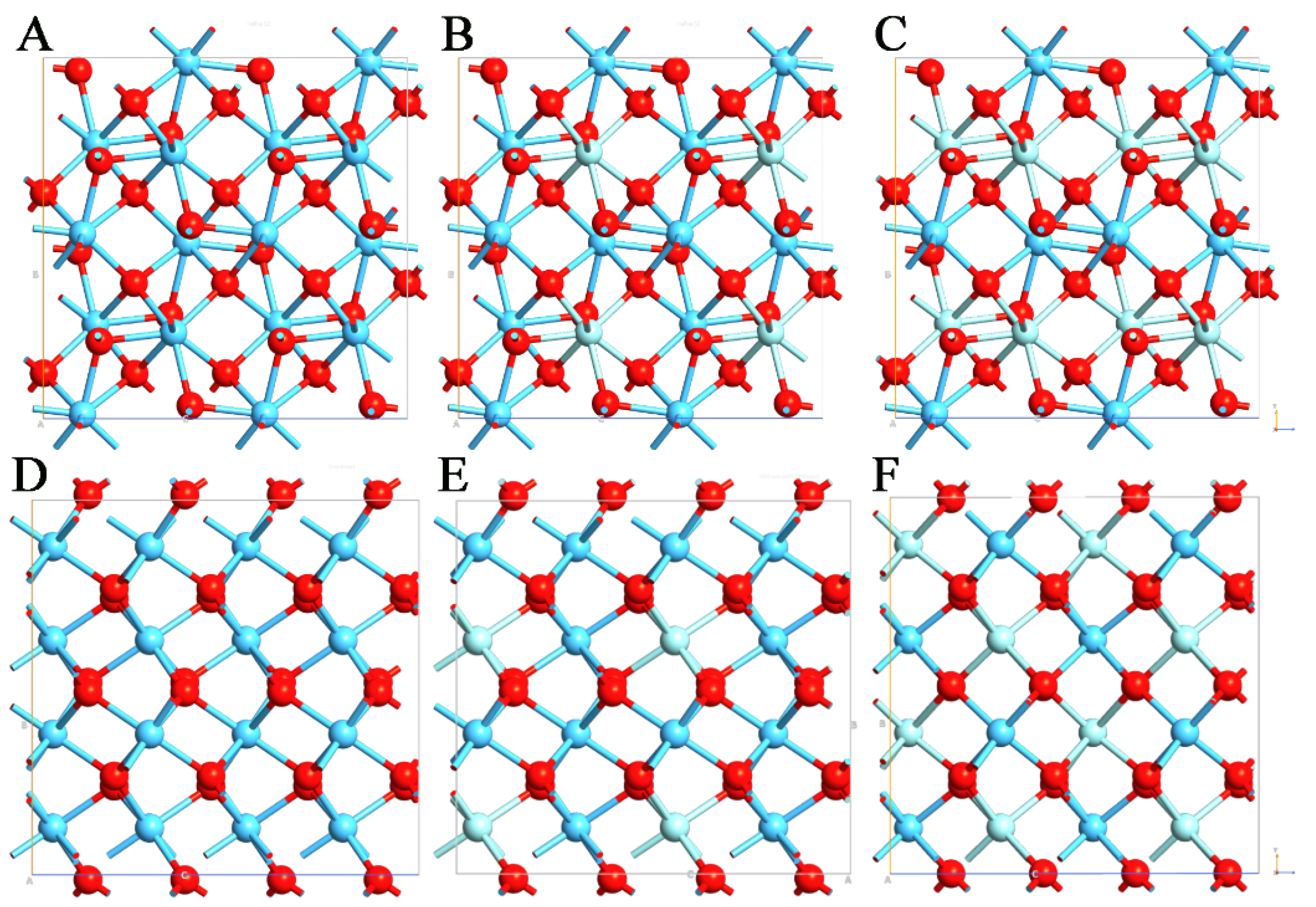
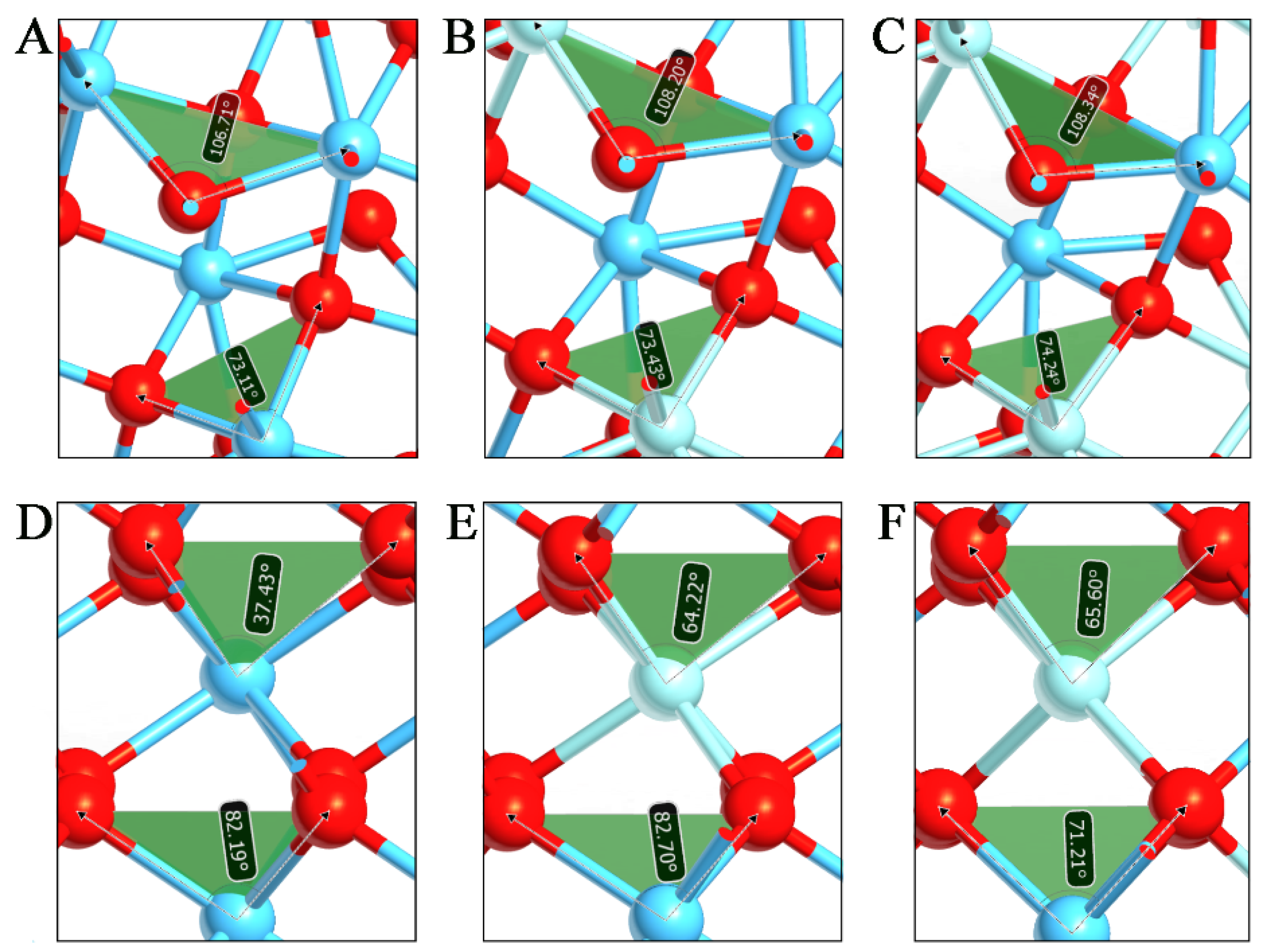
3.1. Effects of Zr on Lattice Parameters
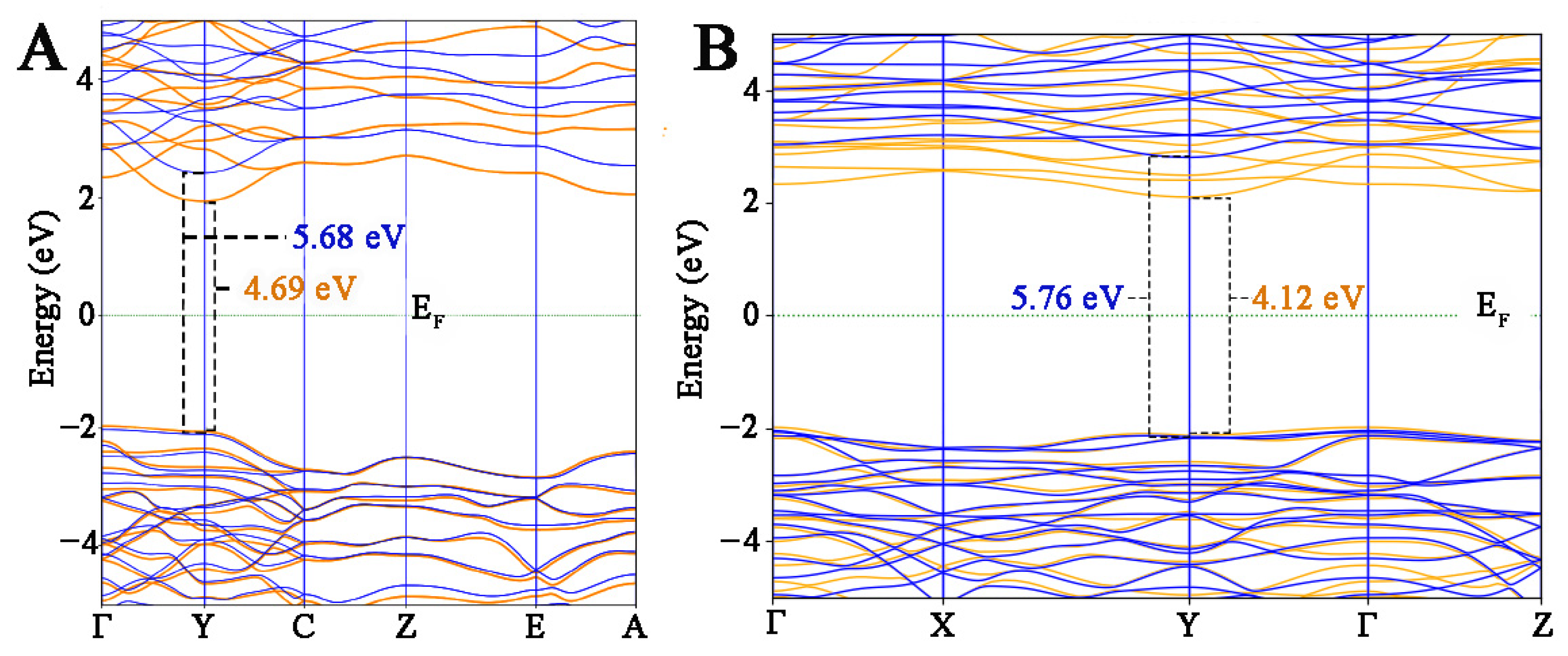
3.2. DFT + U Calibration
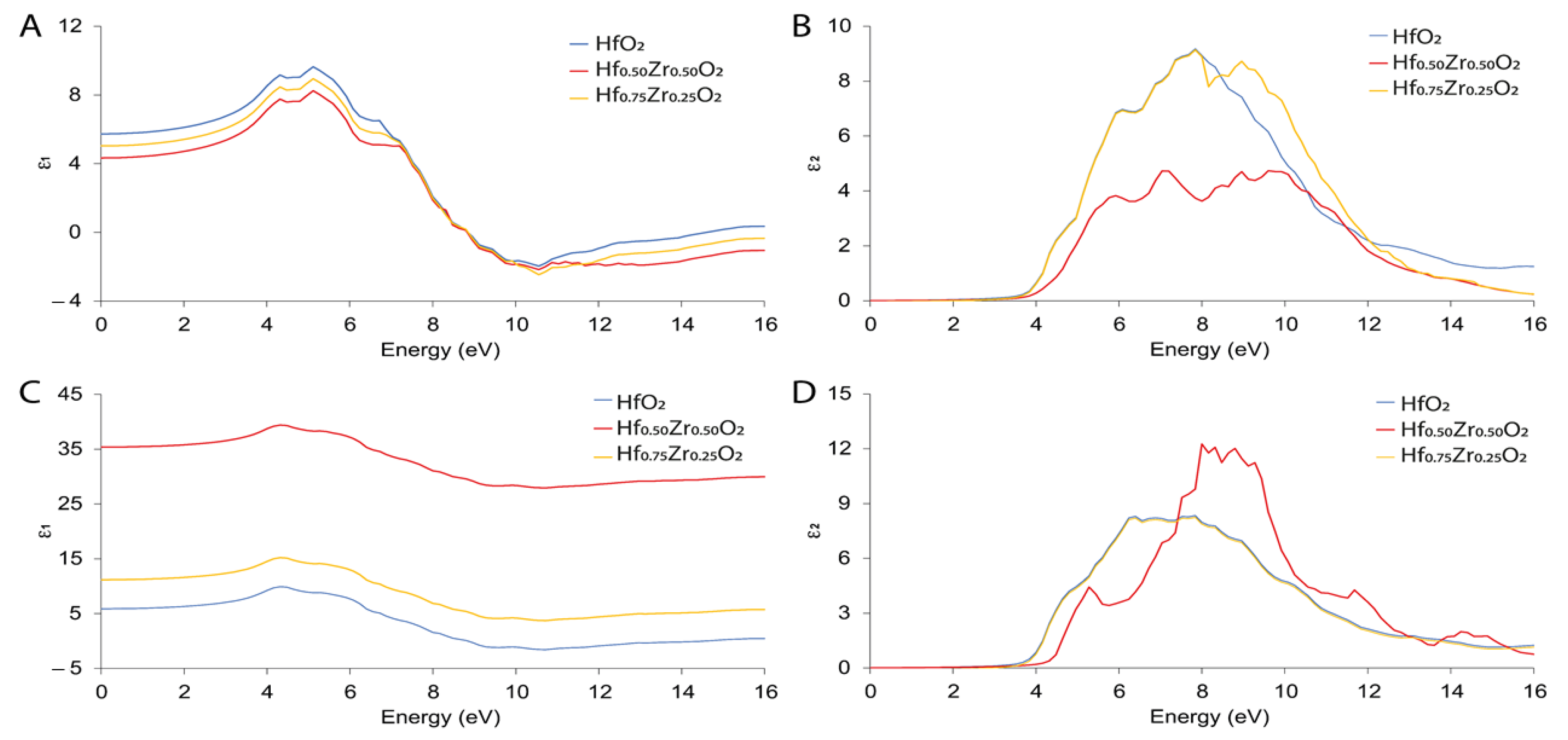
3.3. Electrical and Optical Properties Calculation
3.4. Oxygen Vacancies Effects
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Böscke, T.S.; Müller, J.; Bräuhaus, D.; Schröder, U.; Böttger, U. Ferroelectricity in Hafnium Oxide Thin Films. Appl. Phys. Lett. 2011, 99, 102903. [Google Scholar] [CrossRef]
- Choi, J.H.; Mao, Y.; Chang, J.P. Development of Hafnium Based High-k Materials—A Review. Mater. Sci. Eng. R Rep. 2011, 72, 97–136. [Google Scholar] [CrossRef]
- Zhou, D.; Müller, J.; Xu, J.; Knebel, S.; Bräuhaus, D.; Schröder, U. Insights into Electrical Characteristics of Silicon Doped Hafnium Oxide Ferroelectric Thin Films. Appl. Phys. Lett. 2012, 100, 082905. [Google Scholar] [CrossRef]
- Robertson, J. High Dielectric Constant Gate Oxides for Metal Oxide Si Transistors. Rep. Prog. Phys. 2006, 69, 327–396. [Google Scholar] [CrossRef]
- Dragoman, M.; Modreanu, M.; Povey, I.M.; Dinescu, A.; Dragoman, D.; di Donato, A.; Pavoni, E.; Farina, M. Wafer-Scale Very Large Memory Windows in Graphene Monolayer/HfZrO Ferroelectric Capacitors. Nanotechnology 2018, 29, 425204. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Mueller, J.; Singh, A.; Riedel, S.; Sundqvist, J.; Schroeder, U.; Mikolajick, T. Incipient Ferroelectricity in Al-Doped HfO2 Thin Films. Adv. Funct. Mater. 2012, 22, 2412–2417. [Google Scholar] [CrossRef]
- Olsen, T.; Schröder, U.; Müller, S.; Krause, A.; Martin, D.; Singh, A.; Müller, J.; Geidel, M.; Mikolajick, T. Co-Sputtering Yttrium into Hafnium Oxide Thin Films to Produce Ferroelectric Properties. Appl. Phys. Lett. 2012, 101, 082905. [Google Scholar] [CrossRef]
- Laudadio, E.; Stipa, P.; Pierantoni, L.; Mencarelli, D. Phase Properties of Different HfO2 Polymorphs: A DFT-Based Study. Crystals 2022, 12, 90. [Google Scholar] [CrossRef]
- Zhao, X.; Vanderbilt, D. First-Principles Study of Structural, Vibrational, and Lattice Dielectric Properties of Hafnium Oxide. Phys. Rev. B 2002, 65, 233106. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P. Density Functional Theory and the Band Gap Problem. Int. J. Quantum Chem. 1985, 28, 497–523. [Google Scholar] [CrossRef]
- Wang, L.; Maxisch, T.; Ceder, G. Oxidation Energies of Transition Metal Oxides within the GGA+U Framework. Phys. Rev. B 2006, 73, 195107. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Meng, S.; Niu, J.; Lu, H. Electronic Structures and Optical Properties of Monoclinic ZrO2 Studied by First-Principles Local Density Approximation + U Approach. J. Adv. Ceram. 2017, 6, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Coury, M.E.A.; Dudarev, S.L.; Foulkes, W.M.C.; Horsfield, A.P.; Ma, P.W.; Spencer, J.S. Hubbard-like Hamiltonians for Interacting Electrons in s, p, and d Orbitals. Phys. Rev. B 2016, 93, 075101. [Google Scholar] [CrossRef] [Green Version]
- Smidstrup, S.; Markussen, T.; Vancraeyveld, P.; Wellendorff, J.; Schneider, J.; Gunst, T.; Verstichel, B.; Stradi, D.; Khomyakov, P.A.; Vej-Hansen, U.G.; et al. QuantumATK: An Integrated Platform of Electronic and Atomic-Scale Modelling Tools. J. Phys. Condens. Matter 2020, 32, 015901. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA Method for Ab Initio Order-N Materials Simulation. J. Phys. Condens. Matter 2002, 14, 2745. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Van Setten, M.J.; Giantomassi, M.; Bousquet, E.; Verstraete, M.J.; Hamann, D.R.; Gonze, X.; Rignanese, G.M. The PSEUDODOJO: Training and Grading a 85 Element Optimized Norm-Conserving Pseudopotential Table. Comput. Phys. Commun. 2018, 226, 39–54. [Google Scholar] [CrossRef] [Green Version]
- King-Smith, R.D.; Vanderbilt, D. Theory of Polarization of Crystalline Solids. Phys. Rev. B 1993, 47, 15–1993. [Google Scholar] [CrossRef]
- Fan, S.-T.; Chen, Y.-W.; Liu, C.W. Strain Effect on the Stability in Ferroelectric HfO2 Simulated by First-Principles Calculations. J. Phys. D Appl. Phys. 2020, 53, 23LT01. [Google Scholar] [CrossRef]
- Adams, D.M.; Leonard, S.; Russell, D.R.; Cernik, R.J. X-ray Diffraction Study of Hafnia Under High Pressure Using Synchrotron Radiation. J. Phys. Chem. Solids 1991, 52, 1181–1186. [Google Scholar] [CrossRef]
- Sang, X.; Grimley, E.D.; Schenk, T.; Schroeder, U.; Lebeau, J.M. On the Structural Origins of Ferroelectricity in HfO2 Thin Films. Appl. Phys. Lett. 2015, 106, 162905. [Google Scholar] [CrossRef]
- Senami, M.; Tsuchida, Y.; Fukushima, A.; Ikeda, Y.; Tachibana, A. Local Dielectric Property of Cubic, Tetragonal, and Monoclinic Hafnium Oxides. Jpn. J. Appl. Phys. 2012, 51, 031101. [Google Scholar] [CrossRef]
- Muhammady, S.; Kurniawan, Y.; Suryana; Azizah, N.; Darma, Y. The Effect of Co-Existing Cations on Optical Conductivity and Absorption in Hf0.5Zr0.5O2 System: A First-Principles Study. J. Phys. Conf. Ser. 2019, 1153, 012082. [Google Scholar] [CrossRef]
- Alam, M.N.K.; Clima, S.; O’sullivan, B.J.; Kaczer, B.; Pourtois, G.; Heyns, M.; van Houdt, J. First Principles Investigation of Charge Transition Levels in Monoclinic, Orthorhombic, Tetragonal, and Cubic Crystallographic Phases of HfO2. J. Appl. Phys. 2021, 129, 084102. [Google Scholar] [CrossRef]
- Kim, S.J.; Mohan, J.; Kim, H.S.; Lee, J.; Hwang, S.M.; Narayan, D.; Lee, J.G.; Young, C.D.; Colombo, L.; Goodman, G.; et al. Effect of Hydrogen Derived from Oxygen Source on Low-Temperature Ferroelectric TiN/Hf0.5Zr0.5O2/TiN Capacitors. Appl. Phys. Lett. 2019, 115, 182901. [Google Scholar] [CrossRef]
- Park, M.H.; Lee, Y.H.; Kim, H.J.; Schenk, T.; Lee, W.; Kim, K.; Fengler, F.P.G.; Mikolajick, T.; Schroeder, U.; Hwang, C.S. Surface and Grain Boundary Energy as the Key Enabler of Ferroelectricity in Nanoscale Hafnia-Zirconia: A Comparison of Model and Experiment. Nanoscale 2017, 9, 9973–9986. [Google Scholar] [CrossRef] [Green Version]
- Kaczkowski, J.; Pugaczowa-Michalska, M.; Płowaś-Korus, I. Comparative Density Functional Studies of Pristine and Doped Bismuth Ferrite Polymorphs by GGA+Uandmeta-GGA SCAN+U. Phys. Chem. Chem. Phys. 2021, 23, 8571–8584. [Google Scholar] [CrossRef]
- Lim, S.G.; Kriventsov, S.; Jackson, T.N.; Haeni, J.H.; Schlom, D.G.; Balbashov, A.M.; Uecker, R.; Reiche, P.; Freeouf, J.L.; Lucovsky, G. Dielectric Functions and Optical Bandgaps of High- K Dielectrics for Metal-Oxide-Semiconductor Field-Effect Transistors by Far Ultraviolet Spectroscopic Ellipsometry. J. Appl. Phys. 2002, 91, 4500–4505. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO-MO Molecular Wave Functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Kim, H.J.; Moon, T.; Kim, K.D.; Hyun, S.D.; Park, H.W.; Lee, Y.B.; Park, M.H.; Hwang, C.S. Preparation and Characterization of Ferroelectric Hf0.5Zr0.5O2 Thin Films Grown by Reactive Sputtering. Nanotechnology 2017, 28, 305703. [Google Scholar] [CrossRef]


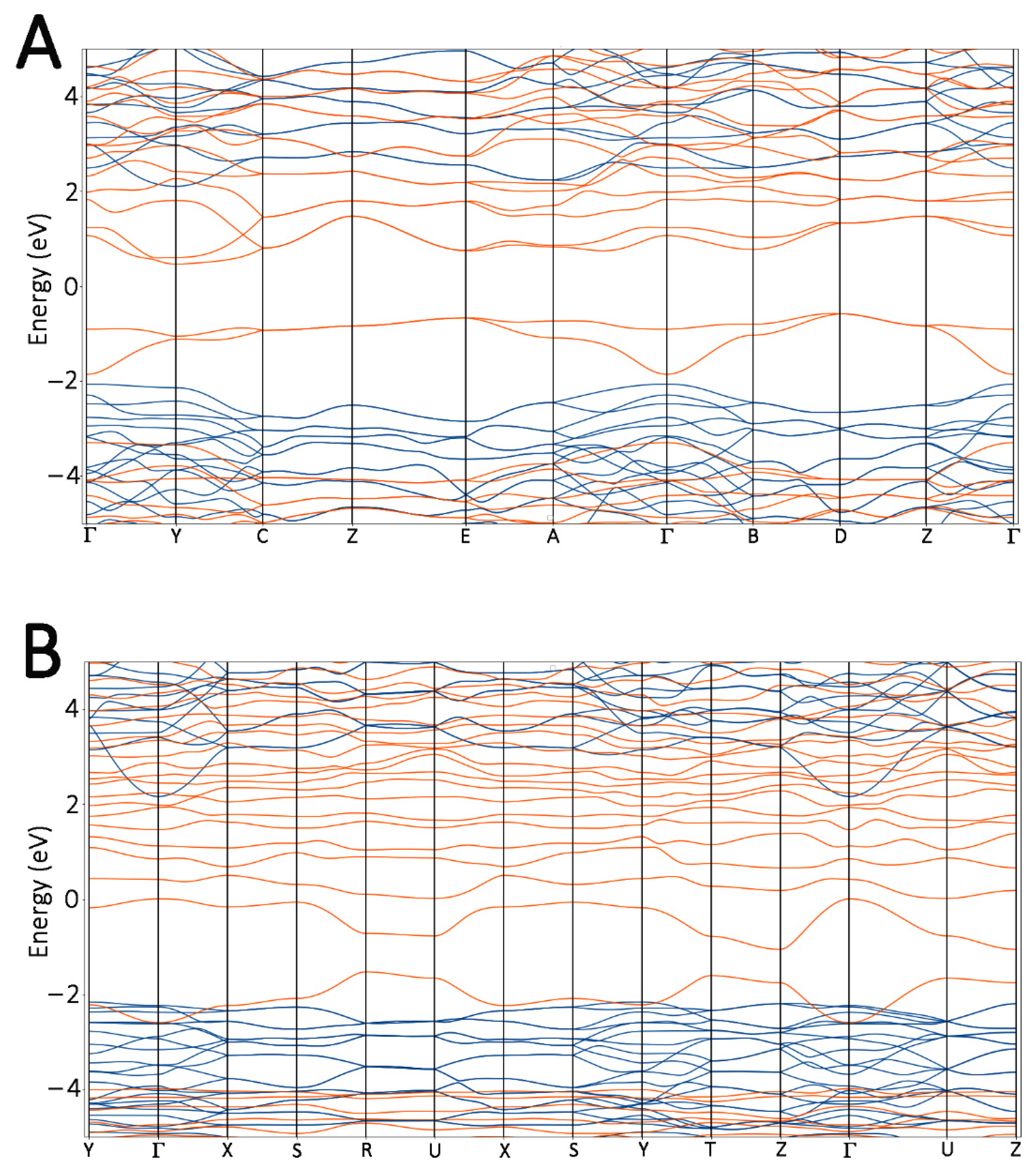

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | HfO2 | Hf0.75Zr0.25 O2 | Hf0.5Zr0.5 O2 | Comparison HfO2 |
|---|---|---|---|---|
| m–P21/c | a = 5.068 | a = 5.065 | a = 5.064 | a = 5.07 |
| b = 5.135 | b = 5.134 | b = 5.135 | b = 5.14 | |
| c = 5.292 | c = 5.290 | c = 5.289 | c = 5.29 | |
| o–Pca21 | a = 5.231 | a = 5.228 | a = 5.229 | a = 5.23 |
| b = 5.008 | b = 4.988 | b = 5.001 | b = 5.00 | |
| c = 5.052 | c = 5.052 | c = 5.031 | c = 5.05 |
| Model | Species | Total (s + p + d) | Charge (a.u.) |
|---|---|---|---|
| P21/c HfO2 | Hf | 2.320 | 1.680 |
| O | 6.840 | −0.840 | |
| P21/c Hf0.75Zr0.25O2 | Hf | 2.723 | 1.233 |
| Zr | 2.323 | 1.023 | |
| O | 6.826 | −0.826 | |
| P21/c Hf0.5Zr0.5O2 | Hf | 3.097 | 0.987 |
| Zr | 2.309 | 0.889 | |
| O | 6.812 | −0.812 | |
| Pca21 HfO2 | Hf | 2.345 | 1.705 |
| O | 6.852 | −0.852 | |
| Pca21 Hf0.75Zr0.25O2 | Hf | 2.387 | 1.797 |
| Zr | 2.121 | 1.531 | |
| O | 6.882 | −0.882 | |
| Pca21 Hf0.5Zr0.5O2 | Hf | 2.601 | 1.931 |
| Zr | 2.365 | 1.765 | |
| O | 6.981 | −0.981 |
| Phase | HfxZryO2 Formation Energy | HfxZryO2–z Formation Energy | Defect Formation Energy |
|---|---|---|---|
| HfO2 P21/c | −11.32 | −11.15 | 0.17 |
| HfO2 Pca21 | −9.64 | −9.27 | 0.37 |
| Hf0.75Zr0.25O2 P21/c | −11.31 | −11.13 | 0.18 |
| Hf0.75Zr0.25O2 Pca21 | −10.08 | −9.59 | 0.49 |
| Hf0.5Zr0.5O2 P21/c | −11.33 | −11.12 | 0.21 |
| Hf0.5Zr0.5O2 Pca21 | −10.78 | −9.96 | 0.82 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavoni, E.; Mohebbi, E.; Stipa, P.; Mencarelli, D.; Pierantoni, L.; Laudadio, E. The Role of Zr on Monoclinic and Orthorhombic HfxZryO2 Systems: A First-Principles Study. Materials 2022, 15, 4175. https://doi.org/10.3390/ma15124175
Pavoni E, Mohebbi E, Stipa P, Mencarelli D, Pierantoni L, Laudadio E. The Role of Zr on Monoclinic and Orthorhombic HfxZryO2 Systems: A First-Principles Study. Materials. 2022; 15(12):4175. https://doi.org/10.3390/ma15124175
Chicago/Turabian StylePavoni, Eleonora, Elaheh Mohebbi, Pierluigi Stipa, Davide Mencarelli, Luca Pierantoni, and Emiliano Laudadio. 2022. "The Role of Zr on Monoclinic and Orthorhombic HfxZryO2 Systems: A First-Principles Study" Materials 15, no. 12: 4175. https://doi.org/10.3390/ma15124175
APA StylePavoni, E., Mohebbi, E., Stipa, P., Mencarelli, D., Pierantoni, L., & Laudadio, E. (2022). The Role of Zr on Monoclinic and Orthorhombic HfxZryO2 Systems: A First-Principles Study. Materials, 15(12), 4175. https://doi.org/10.3390/ma15124175