
Structural and Electronic Properties of SnO Downscaled to Monolayer


Abstract
:1. Introduction
2. Methodology
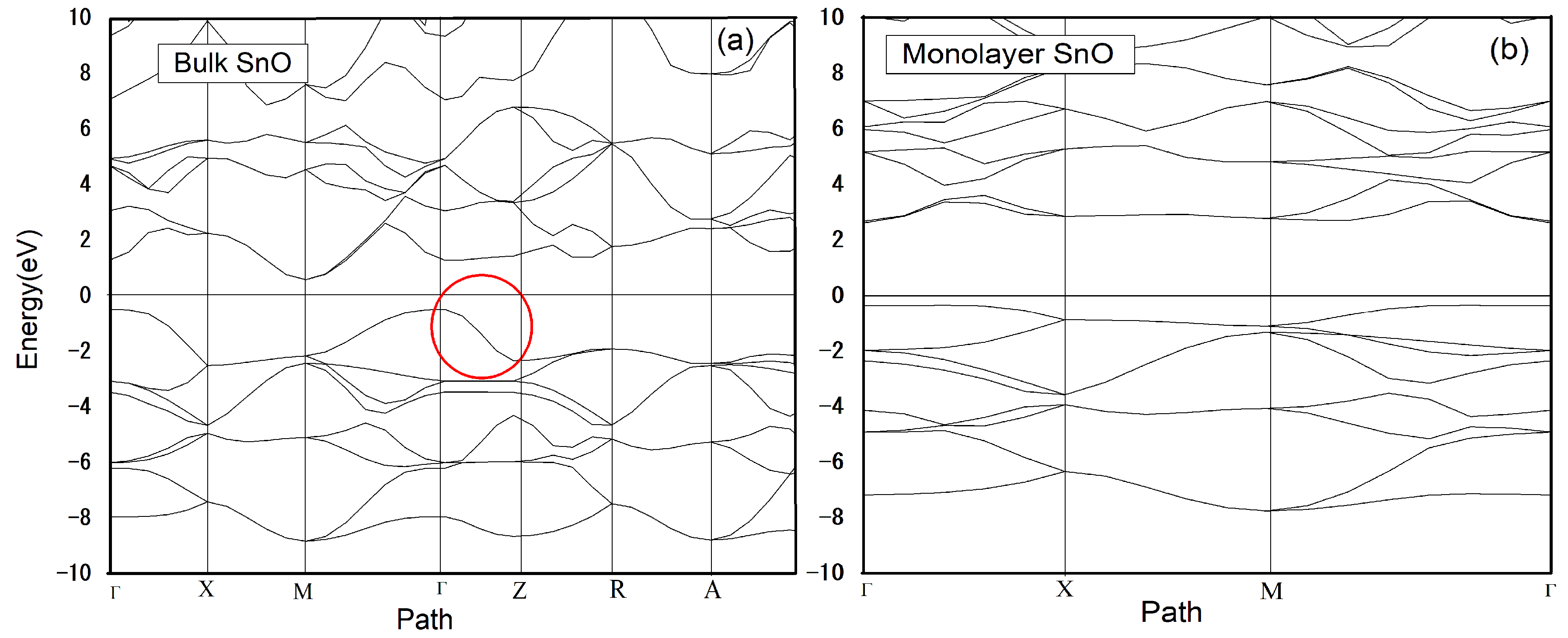
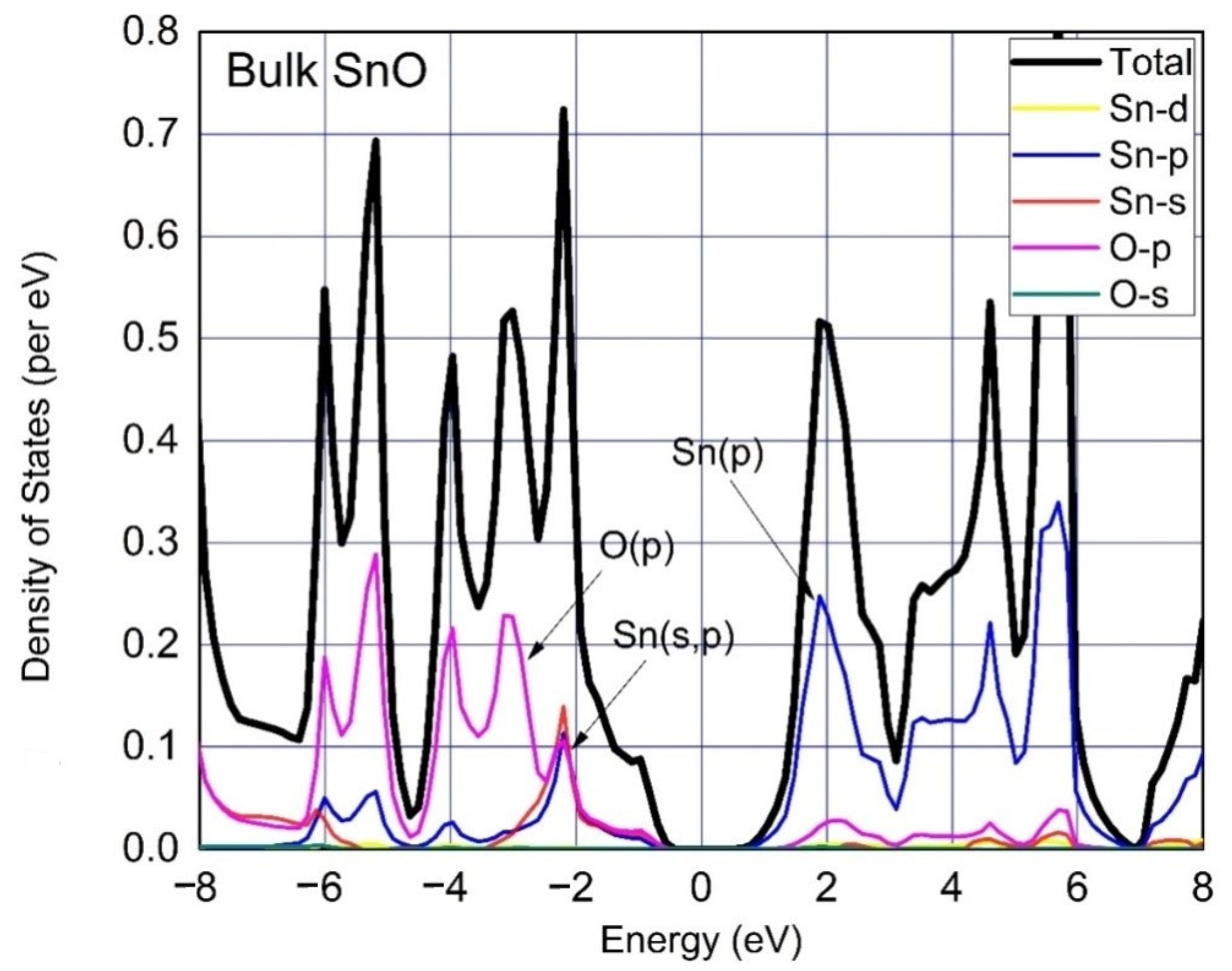
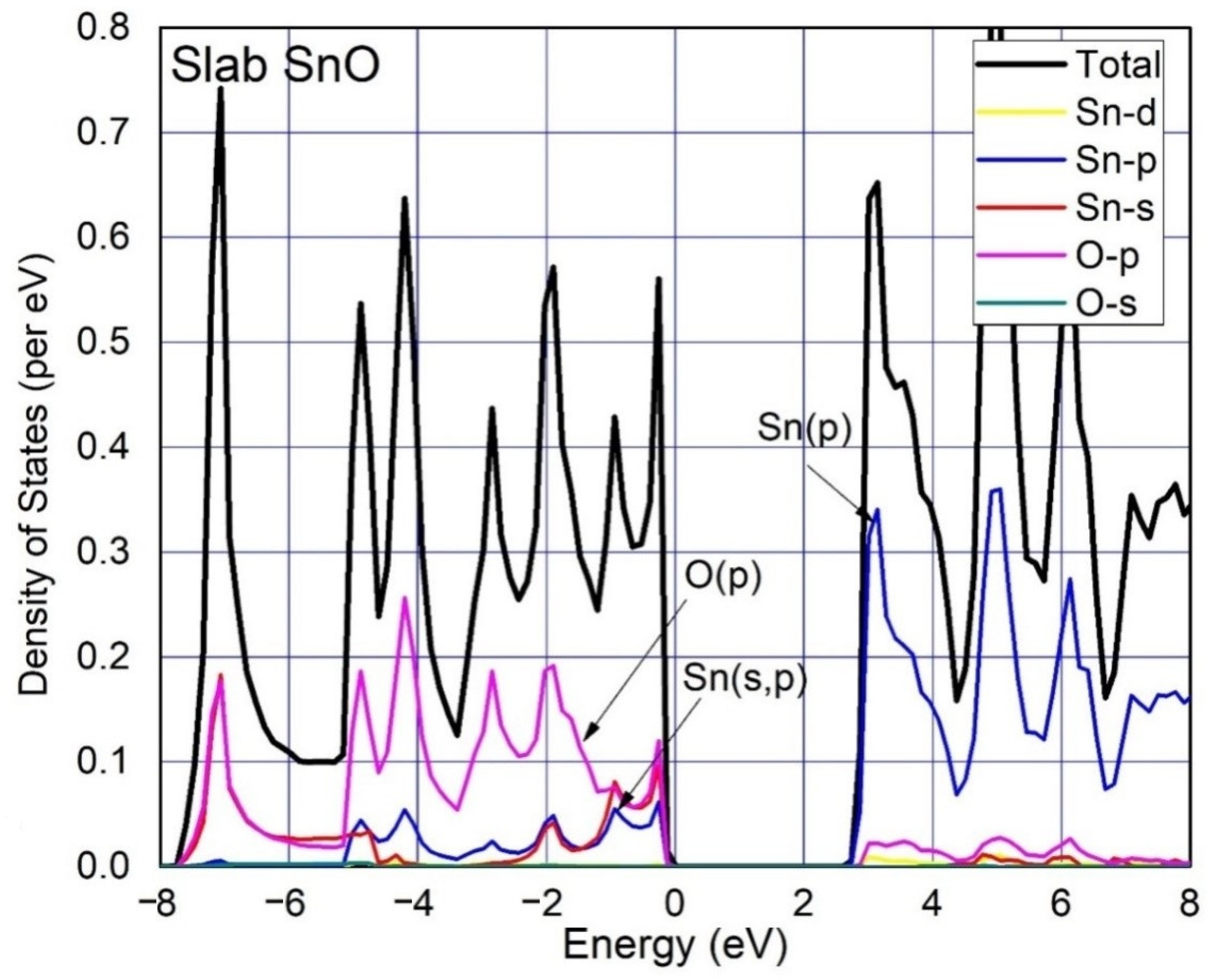
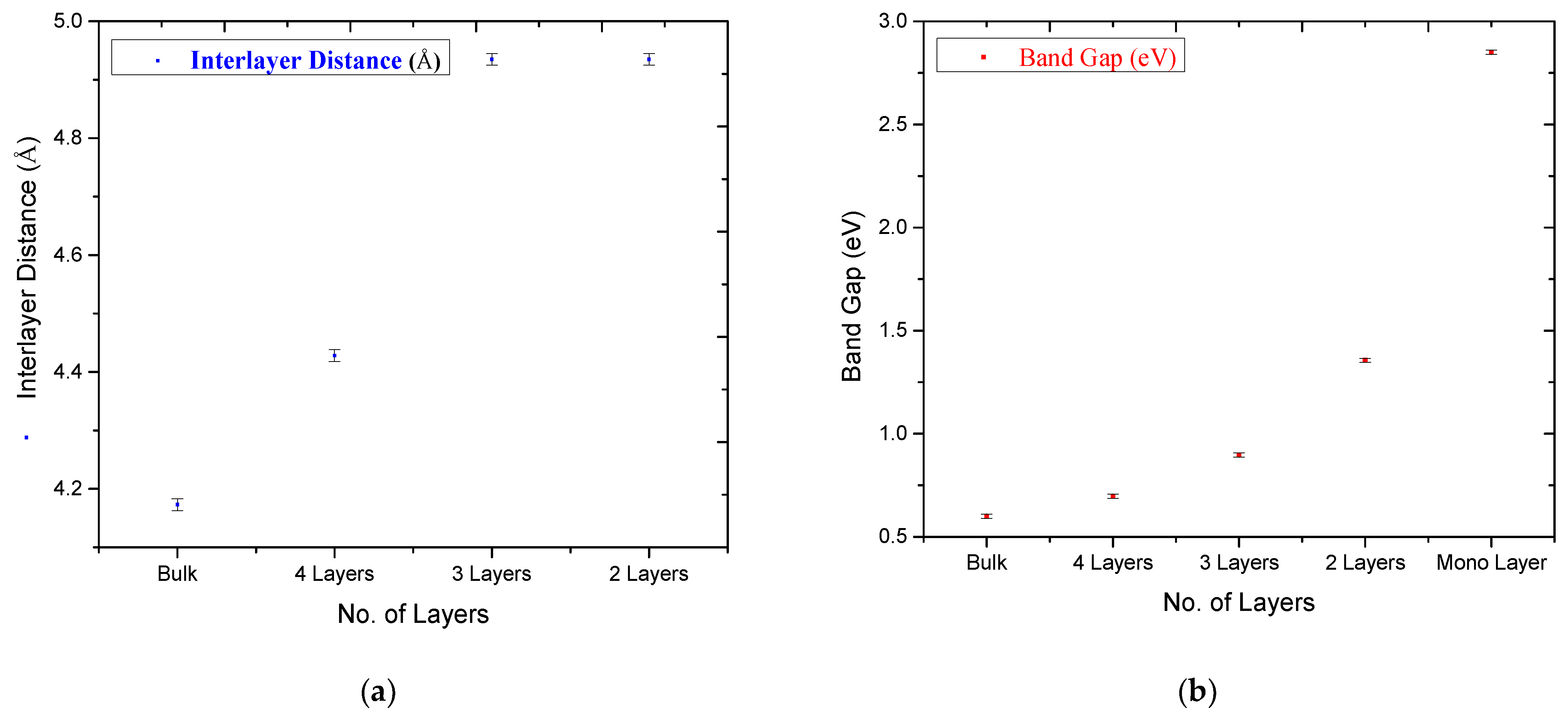
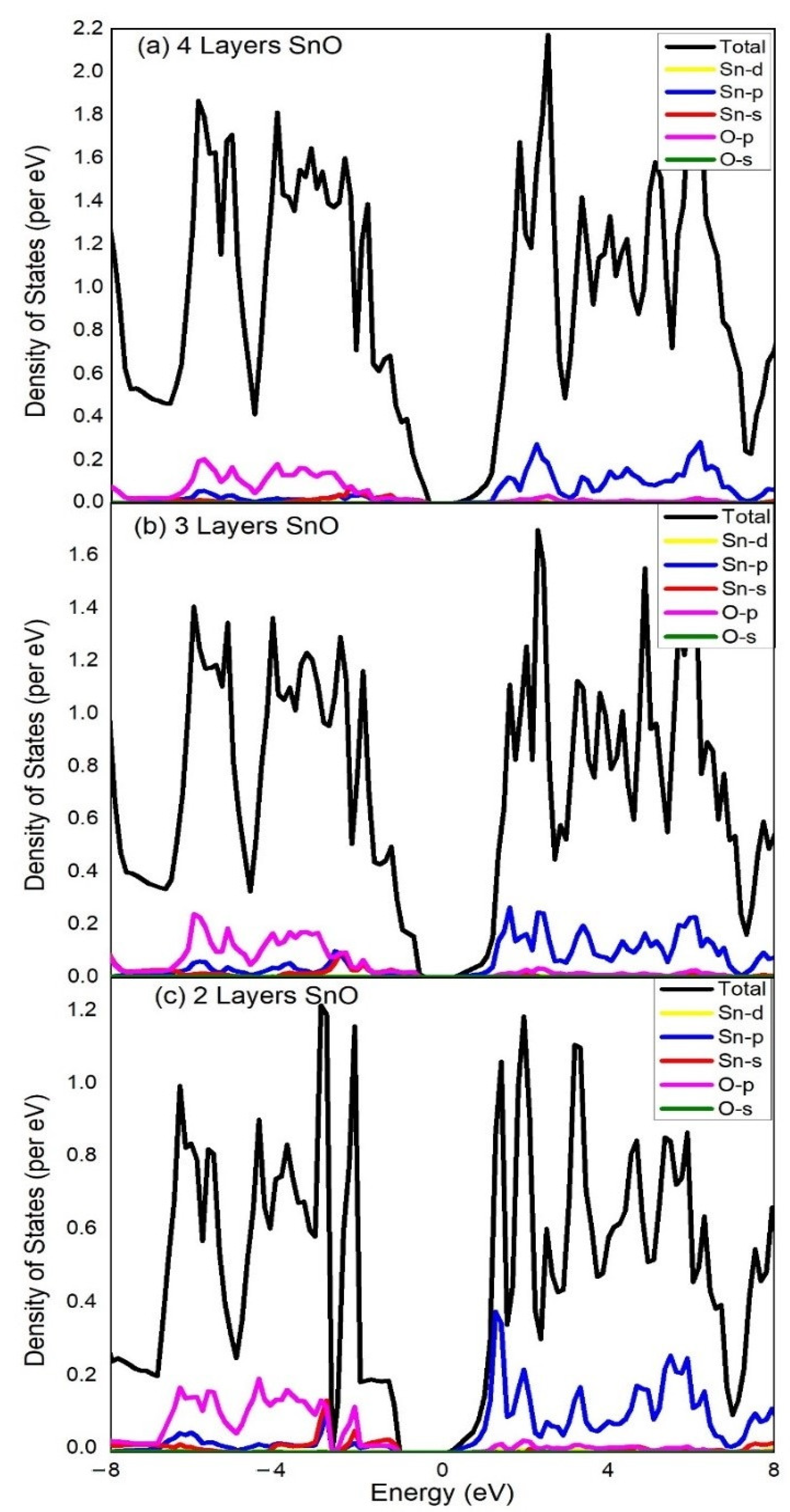
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Daeneke, T.; Atkin, P.; Orrell-Trigg, R.; Zavabeti, A.; Ahmed, T.; Walia, S.; Liu, M.; Tachibana, Y.; Javaid, M.; Greentree, A.D. Wafer-scale synthesis of semiconducting SnO monolayers from interfacial oxide layers of metallic liquid tin. ACS Nano 2017, 11, 10974–10983. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Guan, L. Tailoring the electronic and magnetic properties of monolayer SnO by B, C, N, O and F adatoms. Sci. Rep. 2017, 7, 44568. [Google Scholar] [CrossRef] [PubMed]
- Oba, F.; Choi, M.; Togo, A.; Seko, A.; Tanaka, I. Native defects in oxide semiconductors: A density functional approach. J. Phys. Condens. Matter 2010, 22, 384211. [Google Scholar] [CrossRef] [PubMed]
- Tingting, S.; Fuchun, Z.; Weihu, Z. Density functional theory study on the electronic structure and optical properties of SnO2. Rare Met. Mater. Eng. 2015, 44, 2409–2414. [Google Scholar] [CrossRef]
- Govaerts, K.; Saniz, R.; Partoens, B.; Lamoen, D. van der Waals bonding and the quasiparticle band structure of SnO from first principles. Phys. Rev. B 2013, 87, 235210. [Google Scholar] [CrossRef]
- Hosono, H. Recent progress in transparent oxide semiconductors: Materials and device application. Thin Solid Film. 2007, 515, 6000–6014. [Google Scholar] [CrossRef]
- Yanagi, H.; Inoue, S.-i.; Ueda, K.; Kawazoe, H.; Hosono, H.; Hamada, N. Electronic structure and optoelectronic properties of transparent p-type conducting CuAlO2. J. Appl. Phys. 2000, 88, 4159–4163. [Google Scholar] [CrossRef]
- Kamiya, T.; Hosono, H. Electronic structures and device applications of transparent oxide semiconductors: What is the real merit of oxide semiconductors? Int. J. Appl. Ceram. Technol. 2005, 2, 285–294. [Google Scholar] [CrossRef]
- Farooq, M.H.; Hussain, R.; Aslam, I.; Tanveer, M.; Shah, M.; Iqbal, M.Z. Fabrication, characterization and magnetic properties of Mn-doped SnO nanostructures via hydrothermal method. Mater. Lett. 2014, 131, 350–353. [Google Scholar] [CrossRef]
- Siddiq, N.A.; Chong, W.Y.; Yap, Y.K.; Pramono, Y.H.; Ahmad, H. Tin (IV) oxide nanoparticles as a saturable absorber for a Q-switched erbium-doped fiber laser. Laser Phys. 2018, 28, 125104. [Google Scholar] [CrossRef]
- Wang, Z.; Nayak, P.K.; Caraveo-Frescas, J.A.; Alshareef, H.N. Recent developments in p-Type oxide semiconductor materials and devices. Adv. Mater. 2016, 28, 3831–3892. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.P.; Scanlon, D.O.; Parker, S.C.; Watson, G.W. Tin monoxide: Structural prediction from first principles calculations with van der waals corrections. J. Phys. Chem. C 2011, 115, 19916–19924. [Google Scholar] [CrossRef]
- Quackenbush, N.; Allen, J.; Scanlon, D.; Sallis, S.; Hewlett, J.; Nandur, A.; Chen, B.; Smith, K.; Weiland, C.; Fischer, D. Origin of the bipolar doping behavior of SnO from X-ray spectroscopy and density functional theory. Chem. Mater. 2013, 25, 3114–3123. [Google Scholar] [CrossRef]
- Eqbal, E.; Anila, E. Properties of transparent conducting tin monoxide (SnO) thin films prepared by chemical spray pyrolysis method. Phys. B Condens. Matter 2018, 528, 60–65. [Google Scholar] [CrossRef]
- Qu, Y.; Yang, J.; Li, Y.; Zhang, J.; Wang, Q.; Song, A.; Xin, Q. Organic and inorganic passivation of p-type SnO thin-film transistors with different active layer thicknesses. Semicond. Sci. Technol. 2018, 33, 075001. [Google Scholar] [CrossRef]
- Dolabdjian, K.; Görne, A.L.; Dronskowski, R.; Ströbele, M.; Meyer, H.-J. Tin (II) oxide carbodiimide and its relationship to SnO. Dalton Trans. 2018, 47, 13378–13383. [Google Scholar] [CrossRef]
- Barros, R.; Saji, K.J.; Waerenborgh, J.C.; Barquinha, P.; Pereira, L.; Carlos, E.; Martins, R.; Fortunato, E. Role of structure and composition on the performances of p-type tin oxide thin-film transistors processed at low-temperatures. Nanomaterials 2019, 9, 320. [Google Scholar] [CrossRef] [PubMed]
- Wahila, M.J.; Butler, K.T.; Lebens-Higgins, Z.W.; Hendon, C.H.; Nandur, A.S.; Treharne, R.E.; Quackenbush, N.F.; Sallis, S.; Mason, K.; Paik, H. Lone-pair stabilization in transparent amorphous tin oxides: A potential route to p-type conduction pathways. Chem. Mater. 2016, 28, 4706–4713. [Google Scholar] [CrossRef]
- Wang, J.; Umezawa, N.; Hosono, H. Mixed valence tin oxides as novel van der Waals materials: Theoretical predictions and potential applications. Adv. Energy Mater. 2016, 6, 1501190. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, H.; Dion, M.; Jacobson, N.; Schröder, E.; Hyldgaard, P.; Simak, S.; Langreth, D.C.; Lundqvist, B.I. Van der Waals density functional for layered structures. Phys. Rev. Lett. 2003, 91, 126402. [Google Scholar] [CrossRef]
- Varley, J.; Schleife, A.; Janotti, A.; Van de Walle, C. Ambipolar doping in SnO. Appl. Phys. Lett. 2013, 103, 082118. [Google Scholar] [CrossRef]
- Te Velde, G.t.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Zhou, W.; Umezawa, N. Band gap engineering of bulk and nanosheet SnO: An insight into the interlayer Sn–Sn lone pair interactions. Phys. Chem. Chem. Phys. 2015, 17, 17816–17820. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Van Lenthe, E.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1–118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef] [PubMed]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Yang, X.; Liang, H.-J.; Zhao, X.-X.; Yu, H.-Y.; Wang, M.-Y.; Nie, X.-J.; Wu, X.-L. A sandwich nanocomposite composed of commercially available SnO and reduced graphene oxide as advanced anode materials for sodium-ion full batteries. Inorg. Chem. Front. 2021, 8, 396–404. [Google Scholar] [CrossRef]
- Saji, K.J.; Subbaiah, Y.V.; Tian, K.; Tiwari, A. P-type SnO thin films and SnO/ZnO heterostructures for all-oxide electronic and optoelectronic device applications. Thin Solid Film. 2016, 605, 193–201. [Google Scholar] [CrossRef]
- Chakraborty, D.; Berland, K.; Thonhauser, T. Next-Generation Nonlocal van der Waals Density Functional. J. Chem. Theory 2020, 16, 5893–5911. [Google Scholar] [CrossRef]
- Nie, K.; Wang, X.; Mi, W. Magnetic proximity effect induced spin-dependent electronic structure in two-dimensional SnO by half-metallic monolayer CrN ferromagnet. Phys. Chem. Chem. Phys. 2019, 21, 6984–6990. [Google Scholar] [CrossRef] [PubMed]
- Houssa, M.; Iordanidou, K.; Pourtois, G.; Afanas’ev, V.; Stesmans, A. Ferromagnetism in two-dimensional hole-doped SnO. Aip Adv. 2018, 8, 055010. [Google Scholar] [CrossRef]
- Degler, D. Spectroscopic Insights in the Gas Detection Mechanism of Tin Dioxide Based Gas Sensors. Ph.D. Thesis, Eberhard Karls Universität Tübingen, Tübingen, Germany, 2017. [Google Scholar]
- Ehsani, A.; Mahjani, M.G.; Babaei, F.; Mostaanzadeh, H. Physioelectrochemical and DFT investigation of metal oxide/p-type conductive polymer nanoparticles as an efficient catalyst for the electrocatalytic oxidation of methanol. RSC Adv. 2015, 5, 30394–30404. [Google Scholar] [CrossRef]
- Cordero, B.; Gómez, V.; Platero-Prats, A.E.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent radii revisited. Dalton Trans. 2008, 21, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.; Watson, G.W. Influence of the anion on lone pair formation in Sn (II) monochalcogenides: A DFT study. J. Phys. Chem. B 2005, 109, 18868–18875. [Google Scholar] [CrossRef]
- Allen, J.P.; Scanlon, D.O.; Piper, L.F.; Watson, G.W. Understanding the defect chemistry of tin monoxide. J. Mater. Chem. C 2013, 1, 8194–8208. [Google Scholar] [CrossRef]
- Chen, P.-J.; Jeng, H.-T. Phase diagram of the layered oxide SnO: GW and electron-phonon studies. Sci. Rep. 2015, 5, 16359. [Google Scholar] [CrossRef]
- Pawar, V.; Jha, P.K.; Panda, S.; Jha, P.A.; Singh, P. Band-gap engineering in ZnO thin films: A combined experimental and theoretical study. Phys. Rev. Appl. 2018, 9, 054001. [Google Scholar] [CrossRef]
- Mak, K.F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T.F. Atomically thin MoS 2: A new direct-gap semiconductor. Phys. Rev. Lett. 2010, 105, 136805. [Google Scholar] [CrossRef]
- Kuc, A.; Zibouche, N.; Heine, T. Influence of quantum confinement on the electronic structure of the transition metal sulfide T S 2. Phys. Rev. B 2011, 83, 245213. [Google Scholar] [CrossRef]
- Kang, J.; Zhang, L.; Wei, S.-H. A unified understanding of the thickness-dependent bandgap transition in hexagonal two-dimensional semiconductors. J. Phys. Chem. Lett. 2016, 7, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.V.; Lavrentyev, A.; Thuan, D.V.; Nguyen, C.V.; Khyzhun, O.; Gabrelian, B.; Tran, K.C.; Luong, H.L.; Tung, P.D.; Pham, K.D. Electronic properties and optical behaviors of bulk and monolayer ZrS2: A theoretical investigation. Superlattices Microstruct. 2019, 125, 205–213. [Google Scholar] [CrossRef]
- Heath, J.J.; Costa, M.; Buongiorno-Nardelli, M.; Kuroda, M.A. Role of quantum confinement and interlayer coupling in CrI 3-graphene magnetic tunnel junctions. Phys. Rev. B 2020, 101, 195439. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional | Formation Energy (eV) | Band Gap (eV) |
|---|---|---|
| Experimental | - | 2.5 to 3.3 [12] |
| PBE | −19.87 | 1.544 |
| BP86 | −17.55 | 0.319 |
| BLYP | −15.90 | 0.103 |
| mPBE | −17.54 | 0.185 |
| mPW | −17.52 | 0.24 |
| OLYP | −15.99 | 0.22 |
| OPBE | −17.62 | 0.358 |
| PW91 | −18.99 | 0.274 |
| revPBE | −16.71 | 0.104 |
| RPBE | −16.51 | 0.058 |
| (a) | |||||
| Parameters | PBE: DFT+D3 | PBE [12] | HSE [12] | HSE+optB86-vdW [24] | Exp. [12] |
| a (Å) | 3.859 | 3.860 | 3.794 | 3.847 | 3.801 |
| b (Å) | 3.859 | 3.860 | 3.794 | 3.847 | 3.801 |
| c (Å) | 5.533 | 5.042 | 4.912 | 4.823 | 4.835 |
| Volume (Å)3 | 82.4 | 75.14 | - | - | 69.85 |
| Sn-O (Å) | 2.266 | 2.252 | - | - | 2.222 |
| Sn-Sn (Å) | 3.619 | 3.582 | - | - | 3.539 |
| O-O (Å) | 2.729 | 2.730 | - | - | 2.688 |
| Band Gap (eV) | 0.6 | 0.4 [24] | 0.8 [24] | 1.1 [24] | 0.7 [24] |
| (b) | |||||
| Parameter | PBE: DFT+D3 | PBE [5] | PBE:DFT+D2 [31] | PBE: DFT+D2 [32] | Exp. [12] |
| a (Å) | 3.861 | 3.866 | 3.83 | 3.86 | 3.801 |
| b (Å) | 3.861 | 3.866 | 3.83 | 3.86 | 3.801 |
| Volume (Å)3 | 14.9 | - | - | - | - |
| Sn-O (Å) | 2.266 | - | 1.805 | 2.25 | - |
| Band Gap (eV) | 2.85 | - | 3.281 | 2.7 | 2.5 to 3.3 |
| Parameter | Monolayer | 2 Layers | 3 Layers | 4 Layers | Bulk | |
|---|---|---|---|---|---|---|
| a (Å) | 3.86 | 3.88 | 3.87 | 3.87 | 3.86 | |
| b (Å) | 3.86 | 3.86 | 3.87 | 3.87 | 3.86 | |
| Volume (Å)3 | 14.9 | 15 | 15 | 15 | 82.4 | |
| Intra-layer | ||||||
| Sn-O (Å) | 2.267 | 2.266 | 2.266 | 2.265 | 2.266 | |
| Sn-Sn (Å) | 3.619 | 3.612 | 3.612 | 3.612 | 3.616 | |
| O-O (Å) | 2.731 | 2.74 | 2.74 | 2.74 | 2.73 | |
| Inter-layer | ||||||
| O-O (Å) | - | 4.945 | 4.945 | 4.944 | 4.944 | |
| Sn-Sn (Å) | - | 4.935 | 4.935 | 4.428 | 4.173 | |
| Band Gap (eV) | 2.85 | 1.356 | 0.896 | 0.697 | 0.6 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mubeen, A.; Majid, A.; Alkhedher, M.; Tag-ElDin, E.M.; Bulut, N. Structural and Electronic Properties of SnO Downscaled to Monolayer. Materials 2022, 15, 5578. https://doi.org/10.3390/ma15165578
Mubeen A, Majid A, Alkhedher M, Tag-ElDin EM, Bulut N. Structural and Electronic Properties of SnO Downscaled to Monolayer. Materials. 2022; 15(16):5578. https://doi.org/10.3390/ma15165578
Chicago/Turabian StyleMubeen, Adil, Abdul Majid, Mohammad Alkhedher, ElSayed M. Tag-ElDin, and Niyazi Bulut. 2022. "Structural and Electronic Properties of SnO Downscaled to Monolayer" Materials 15, no. 16: 5578. https://doi.org/10.3390/ma15165578