
First Principle Surface Analysis of YF3 and Isostructural HoF3


Abstract
:
1. Introduction
2. Methodology
2.1. Computational Details
2.2. Choice of Electronic Structure Method
2.3. Surface Energy
3. Results and Discussion
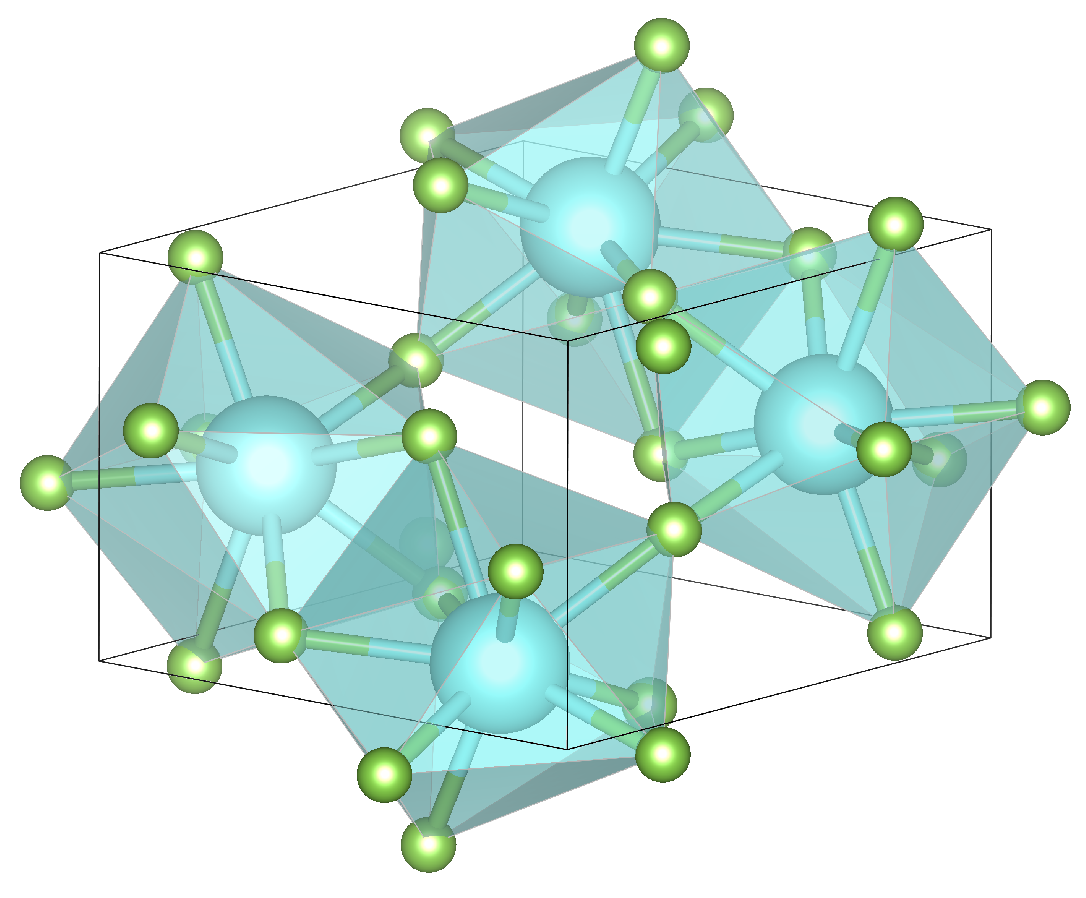
3.1. Bulk Properties
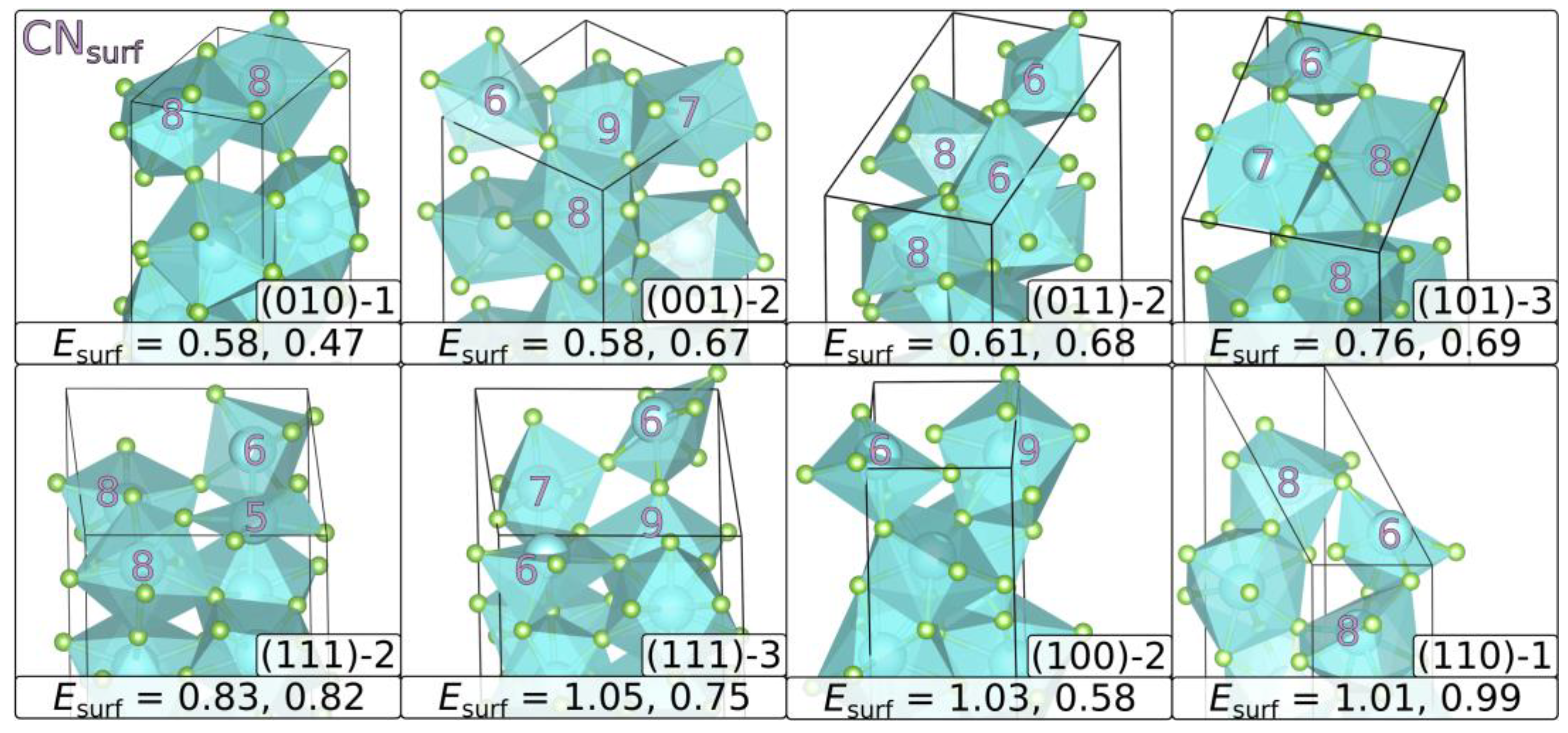
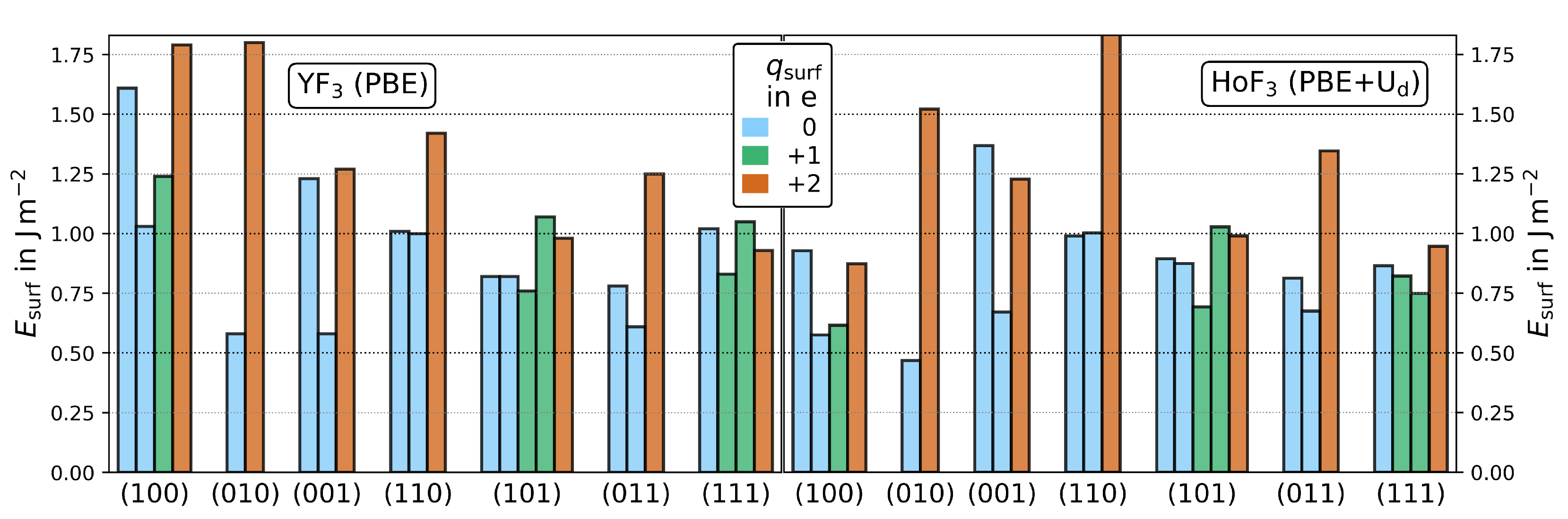
3.2. Surface Energies
3.2.1. Bader Charges
3.2.2. Surface Band Gaps
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Bau, M.; Dulski, P. Comparative study of yttrium and rare-earth element behaviours in fluorine-rich hydrothermal fluids. Contrib. Mineral. Petrol. 1995, 119, 213–223. [Google Scholar] [CrossRef]
- Minuzzi, O.R.R.; Bastos Neto, A.C.; Formoso, M.L.L.; Andrade, S.; Janasi, V.A.; Flores, J.A. Rare earth element and yttrium geochemistry applied to the genetic study of cryolite ore at the Pitinga Mine (Amazon, Brazil). An. Acad. Bras. Cienc. 2008, 80, 719–733. [Google Scholar] [CrossRef]
- Goodenough, K.M.; Schilling, J.; Jonsson, E.; Kalvig, P.; Charles, N.; Tuduri, J.; Deady, E.A.; Sadeghi, M.; Schiellerup, H.; Müller, A.; et al. Europe’s rare earth element resource potential: An overview of REE metallogenetic provinces and their geodynamic setting. Ore Geol. Rev. 2016, 72, 838–856. [Google Scholar] [CrossRef]
- Kaminski, A. Laser Crystals: Their Physics and Properties, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 1990. [Google Scholar]
- Kollia, Z.; Sarantopoulou, E.; Cefalas, A.C.; Nicolaides, C.A.; Naumov, A.K.; Semashko, V.V.; Abdulsabirov, R.Y.; Korableva, S.L.; Dubinskii, M.A. Vacuum-ultraviolet interconfigurational 4f3 → 4f25d absorption and emission studies of the Nd3+ ion in KYF, YF, and YLF crystal hosts. J. Opt. Soc. Am. B 1995, 12, 782–785. [Google Scholar] [CrossRef]
- Lage, M.M.; Righi, A.; Matinaga, F.M.; Gesland, J.Y.; Moreira, R.L. Raman-spectroscopic study of lanthanide trifluorides with the - YF3 structure. J. Phys. Condens. Matter 2004, 16, 3207–3218. [Google Scholar] [CrossRef]
- Atencio, D.; Bastos Neto, A.C.; Pereira, V.P.; Ferron, J.T.M.M.; Hoshino, M.; Moriyama, T.; Watanabe, Y.; Miyawaki, R.; Coutinho, J.M.V.; Andrade, M.B.; et al. Waimirite-(Y), orthorhombic YF3, a new mineral from the Pitinga mine, Presidente Figueiredo, Amazonas, Brazil and from Jabal Tawlah, Saudi Arabia: Description and crystal structure. Mineral. Mag. 2015, 79, 767–780. [Google Scholar] [CrossRef]
- Sarantopoulou, E.; Kollia, Z.; Cefalas, A.C. YF3:Nd3+, Pr3+, Gd3+ wide band gap crystals as optical materials for 157-nm photolithography. Opt. Mater. 2001, 18, 23–26. [Google Scholar] [CrossRef]
- O’Keeffe, M. Ionic Conductivity of Yttrium Fluoride and Lutetium Fluoride. Science 1973, 180, 1276–1277. [Google Scholar] [CrossRef]
- Pogorenko, Y.V.; Pshenychnyi, R.M.; Lutsyk, V.I.; Omel’chuk, A.O. Transport Properties of Aliovalent Substitution Solid Solutions of the System (1-x)PbF2-xYF3-SnF2. IOP Conf. Ser. Mater. Sci. Eng. 2017, 175, 012039. [Google Scholar] [CrossRef]
- Fedorov, P.P.; Sorokin, N.I. Stabilization of the α-YF3 structure type by isomorphous substitutions. Inorg. Mater. 2017, 53, 1307–1311. [Google Scholar] [CrossRef]
- Cui, X.; Hu, T.; Wang, J.; Zhang, J.; Zhong, X.; Chen, Y.; Li, X.; Yang, J.; Gao, C. Ionic transportation and dielectric properties of YF3:Eu3+ nanocrystals. Nanomaterials 2018, 8, 995. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Komatsu, Y.; Shimizu, R.; Hitosugi, T. Diffusion of F atoms from fluoride substrates promotes the epitaxial growth of metal fluorides. Appl. Phys. Express 2020, 13, 85507. [Google Scholar] [CrossRef]
- Hoard, R.; Mance, S.; Leber, R.; Dalder, E.; Chaplin, M.; Blair, K.; Nelson, D.; Dyke, D.V. Field enhancement of a 12.5-T magnet using holmium poles. IEEE Trans. Magn. 1985, 21, 448–450. [Google Scholar] [CrossRef]
- González-Mancebo, D.; Becerro, A.I.; Rojas, T.C.; García-Martín, M.L.; de la Fuente, J.M.; Ocaña, M. HoF3 and DyF3 Nanoparticles as Contrast Agents for High-Field Magnetic Resonance Imaging. Part. Part. Syst. Charact. 2017, 34, 1700116. [Google Scholar] [CrossRef]
- Rudzitis, E.; Feder, H.M.; Hubbard, W.N. Fluorine bomb calorimetry. XI. The enthalpy of formation of yttrium trifluoride. J. Phys. Chem. 1965, 69, 2305–2307. [Google Scholar] [CrossRef]
- Spedding, F.H.; Henderson, D.C. High-temperature heat contents and related thermodynamic functions of seven trifluorides of the rare earths: Y, La, Pr, Nd, Gd, Ho, and Lu. J. Chem. Phys. 1971, 54, 2476–2483. [Google Scholar] [CrossRef]
- Loges, A.; Migdisov, A.A.; Wagner, T.; Williams-Jones, A.E.; Markl, G. Fluoride complexation of hafnium under hydrothermal conditions. In Proceedings of the Goldschmidt Conference, Montréal, QC, Canada, 24–29 June 2012; p. 2030. [Google Scholar]
- Loges, A.; Migdisov, A.A.; Wagner, T.; Williams-Jones, A.E.; Markl, G. An experimental study of the aqueous solubility and speciation of Y(III) fluoride at temperatures up to 250C. Geochim. Cosmochim. Acta 2013, 123, 403–415. [Google Scholar] [CrossRef]
- Bau, M. Controls on the fractionation of isovalent trace elements in magmatic and aqueous systems: Evidence from Y/Ho, Zr/Hf, and lanthanide tetrad effect. Contrib. Mineral. Petrol. 1996, 123, 323–333. [Google Scholar] [CrossRef]
- Zalkin, A.; Templeton, D.H. The Crystal Structures of YF3 and Related Compounds. J. Am. Chem. Soc. 1953, 75, 2453–2458. [Google Scholar] [CrossRef]
- Cheetham, A.K.; Norman, N. The Structures of Yttrium and Bismuth Trifluorides by Neutron Diffraction. Acta Chem. Scand. 1974, 28, 55–60. [Google Scholar] [CrossRef]
- Khairulin, R.A.; Stankus, S.V.; Lyapunov, K.M. The Thermal Properties of Holmium Trifluoride at High Temperatures. High Temp. 2000, 38, 149–151. [Google Scholar] [CrossRef]
- Loges, A.; Freie Universität Berlin, Berlin, Germany. Private communication, 2020.
- Vologzhanina, A.V.; Pushkin, D.V.; Serezhkin, V.N. Coordination polyhedra LnFn (Ln = La-Lu) in crystal structures. Russ. J. Inorg. Chem. 2006, 51, 747–758. [Google Scholar] [CrossRef]
- Rotereau, K.; Daniel, P.; Desert, A.; Gesland, J.Y. The high-temperature phase transition in samarium fluoride: Structural and vibrational investigation. J. Phys. Condens. Matter 1998, 10, 1431–1446. [Google Scholar] [CrossRef]
- Zinchenko, V.; Efryushina, N.; Eryomin, O.; Markiv, V.; Belyavina, N.; Mozkova, O.; Zakharenko, M. Synthesis, structure and optical properties of EuF3 film-forming material. J. Alloys Compd. 2002, 347, L1–L3. [Google Scholar] [CrossRef]
- Piotrowski, M.; Murasik, A. Exchange and Dipolar Interactions in TbF3. Phys. Status Solidi A 1985, 89, 571–580. [Google Scholar] [CrossRef]
- Piotrowski, M.; Ptasiewicz-ba̧k, H.; Murasik, A. The crystal structures of HoF3 and TbF3 by neutron diffraction. Phys. Status Solidi A 1979, 55, K163–K166. [Google Scholar] [CrossRef]
- Bukvetskii, B.; Garashina, L. Crystal-chemical investigation of the orthorhombic trifluorides of samarium, holmium, and ytterbium. Russ. J. Coord. Chem. 1977, 3, 1024–1029. [Google Scholar]
- Peterson, J.R.; Cunningham, B.B. Crystal structures and lattice parameters of the compounds of berkelium—IV berkelium trifluoride. J. Inorg. Nucl. Chem. 1968, 30, 1775–1784. [Google Scholar] [CrossRef]
- Cunningham, B.; Ehrlich, P. USAEC Report UCRL-20426; Technical Report; USAEC: Houston, TX, USA, 1970. [Google Scholar]
- Stevenson, J.N.; Peterson, J.R. The trigonal and orthorhombic crystal structures of CfF3 and their temperature relationship. J. Inorg. Nucl. Chem. 1973, 35, 3481–3486. [Google Scholar] [CrossRef]
- Morss, L.R.; Edelstein, N.M.; Fuger, J. (Eds.) The Chemistry of the Actinide and Transactinide Elements, 3rd ed.; Springer International Publishing: Dordrecht, The Netherlands, 2008; p. 1468. [Google Scholar]
- Haschke, J.M.; Hodges, A.E.; Lucas, R.L. Equilibrium and structural properties of the PuH system. J. Less-Common Met. 1987, 133, 155–166. [Google Scholar] [CrossRef]
- Greis, O.; Martinez-Ripoll, M. Darstellung, Temperaturverhalten und Kristallstruktur von BiF3. Z. Anorg. Allg. Chem. 1977, 436, 105–112. [Google Scholar] [CrossRef]
- Deng, Z.; Wei, F.; Wu, Y.; Seshadri, R.; Cheetham, A.K.; Canepa, P. Understanding the Structural and Electronic Properties of Bismuth Trihalides and Related Compounds. Inorg. Chem. 2020, 59, 3377–3386. [Google Scholar] [CrossRef]
- Ye, X.; Chen, J.; Engel, M.; Millan, J.A.; Li, W.; Qi, L.; Xing, G.; Collins, J.E.; Kagan, C.R.; Li, J.; et al. Competition of shape and interaction patchiness for self-assembling nanoplates. Nat. Chem. 2013, 5, 466–473. [Google Scholar] [CrossRef]
- Griffiths, C.L.; Macdonald, J.E.; Williams, R.H. The growth and characterisation of epitaxial insulating HoF3 layers on silicon. Appl. Surf. Sci. 1992, 56–58, 782–788. [Google Scholar] [CrossRef]
- Guo, F.; Li, H.; Zhang, Z.; Meng, S.; Li, D. Synthesis of mesoporous YF3 nanoflowers via solvent extraction route. Mater. Sci. Eng. B 2009, 163, 134–137. [Google Scholar] [CrossRef]
- Wen, C.; Sun, L.; Yan, J.; Liu, Y.; Song, J.; Zhang, Y.; Lian, H.; Kang, Z. Mesoporous rare earth fluoride nanocrystals and their photoluminescence properties. J. Colloid Interface Sci. 2011, 357, 116–120. [Google Scholar] [CrossRef]
- Tian, Y.; Chen, B.; Li, X.; Zhang, J.; Tian, B.; Sun, J.; Cheng, L.; Zhong, H.; Zhong, H.; Hua, R. Solvothermal synthesis and tunable luminescence of Tb3+, Eu3+ codoped YF3 nano- and micro-crystals with uniform morphologies. J. Solid State Chem. 2012, 196, 187–196. [Google Scholar] [CrossRef]
- Chen, J.x.; Wang, X.p.; Wang, L.J.; Yang, X.w.; Yang, Y. White electroluminescence of diamond—HoF3- diamond composite film. J. Lumin. 2020, 224, 117310. [Google Scholar] [CrossRef]
- Nanda, S.S.; Nayak, P.; Goutam, U.K.; Dash, S. Influence of Eu3+ on the Structure and Photophysical Properties in (Y,Gd)F3 Nanophosphors. J. Fluoresc. 2021, 31, 129–139. [Google Scholar] [CrossRef]
- Pandharkar, R.; Becker, C.; Budau, J.; Kaawar, Z.; Paulus, B. A Computational Study of AlF3 and ACF Surfaces. Inorganics 2018, 6, 124. [Google Scholar] [CrossRef]
- Schnellbügel, A.; Anton, R. On background subtraction for quantitative analysis of X-ray photoelectron spectra of rare earth fluorides. Surf. Sci. 2001, 492, 305–314. [Google Scholar] [CrossRef]
- Wulff, G. XXV. Zur Frage der Geschwindigkeit des Wachstums und der Auflösung der Krystallflächen. Z. Kristallogr.-Cryst. Mater. 1901, 34, 449–530. [Google Scholar] [CrossRef]
- Laue, M.V. Der Wulffsche Satz für die Gleidigewichtsform von Kristallen. Z. Kristallogr.-Cryst. Mater. 1943, 105, 124–133. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Norm-conserving and ultrasoft pseudopotentials for first-row and transition elements. J. Phys. Condens. Matter 1994, 6, 8245–8257. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223–16233. [Google Scholar] [CrossRef]
- Evans, D.S.; Baynor, G.V. Lattice spacings in thorium—Yttrium alloys. J. Nucl. Mater. 1960, 2, 209–215. [Google Scholar] [CrossRef]
- Pechan, M.J.; Stassis, C. Magnetic structure of holmium. J. Appl. Phys. 1984, 55, 1900–1902. [Google Scholar] [CrossRef]
- Kerker, G.P. Efficient iteration scheme for self-consistent pseudopotential calculations. Phys. Rev. B 1981, 23, 3082–3084. [Google Scholar] [CrossRef]
- Sun, W.; Ceder, G. Efficient creation and convergence of surface slabs. Surf. Sci. 2013, 617, 53–59. [Google Scholar] [CrossRef]
- Tran, R.; Xu, Z.; Radhakrishnan, B.; Winston, D.; Sun, W.; Persson, K.A.; Ong, S.P. Surface energies of elemental crystals. Sci. Data 2016, 3, 160080. [Google Scholar] [CrossRef]
- Rahm, J.M.; Erhart, P. WulffPack: A Python package for Wulff constructions. J. Open Source Softw. 2020, 5, 1944. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Rogers, E.; Dorenbos, P.; van der Kolk, E. Systematics in the optical and electronic properties of the binary lanthanide halide, chalcogenide and pnictide compounds: An overview. New J. Phys. 2011, 13, 093038. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comp. Mat. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Sanville, E.; Kenny, S.D.; Smith, R.; Henkelman, G. Improved grid-based algorithm for Bader charge allocation. J. Comput. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 84204. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Trinkle, D.R. Accurate and efficient algorithm for Bader charge integration. J. Chem. Phys. 2011, 134, 64111. [Google Scholar] [CrossRef]
- Brown, P.J.; Forsyth, J.B.; Hansen, P.C.; Leask, M.J.M.M.; Ward, R.C.C.C.; Wells, M.R. Neutron diffraction determination of magnetic order in holmium trifluoride, HoF3. J. Phys. Condens. Matter 1990, 2, 4471–4484. [Google Scholar] [CrossRef]
- Boettger, J.C. Nonconvergence of surface energies obtained from thin-film calculations. Phys. Rev. B 1994, 49, 16798–16800. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| YF3 (PBE) | HoF3 (PBE+U/3 eV/4f-in-Core) | |||||||
|---|---|---|---|---|---|---|---|---|
| (Å) | (Å) | (Å) | (Å) | (Å) | (Å) | (Å) | (Å) | |
| calc. | 6.3215 | 6.8059 | 4.3300 | 186.29 | 6.4164 | 6.8796 | 4.3440 | 191.76 |
| exp. [22] | 6.353(3) | 6.850(3) | 4.393(3) | 191.2 | 6.404(3) | 6.875(3) | 4.379(3) | 192.8 |
| 0.032 | 0.044 | 0.063 | 4.9 | 0.012 | 0.005 | 0.035 | 1.0 | |
| 0.5% | 0.6% | 1.4% | 2.6% | 0.2% | 0.1% | 0.8% | 0.5% | |
| exp. [23] | 6.3537(7) | 6.8545(7) | 4.3953(5) | 191.42 | ||||
| 0.0322 | 0.0486 | 0.0653 | 5.13 | — | ||||
| 0.5% | 0.7% | 1.5% | 2.7% | |||||
| CN | () | % | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| () | term. | YF3 | HoF3 | YF3 | HoF3 | YF3 | HoF3 | YF3 | HoF3 | |
| (100) | 1 | 0 | 20 | 24 | 5,9 | 1.61 (2.87) | 0.93 (1.48) | |||
| 2 | 0 | 22 | 26 | 6,9 | 1.03 (2.02) | 0.58 (0.96) | 7% | 25% | ||
| 3 | +1 | 20 | 24 | 5,8 | 1.24 (1.61) | 0.62 (0.68) | ||||
| 4 | +2 | 22 | 26 | 4,7 | 1.79 (2.14) | 0.87 (0.90) | ||||
| (010) | 1 | 0 | 10 | 12 | 8,8 | 0.58 (0.84) | 0.47 (0.49) | 26% | 34% | |
| 2 | +2 | 10 | 12 | 6,6 | 1.80 (2.05) | 1.52 (1.52) | ||||
| (001) | 1 | 0 | 20 | 24 | 5,8,8,9 | 1.23 (2.45) | 1.37 (2.25) | |||
| 2 | 0 | 22 | 26 | 6,7,8,9 | 0.58 (1.39) | 0.67 (1.16) | 10% | 6% | ||
| 3 | +2 | 22 | 26 | 4,5,8,9 | 1.27 (1.70) | 1.23 (1.29) | ||||
| (110) | 1 | 0 | 20 | 24 | 6,8,8 | 1.01 (1.80) | 0.99 (1.59) | 5% | 0% | |
| 2 | 0 | 22 | 26 | 6,8,8 | 1.00 (2.41) | 1.00 (2.18) | ||||
| 3 | +2 | 22 | 26 | 4,6,9 | 4,6,8 | 1.42 (1.73) | 2.09 (1.36) | |||
| (101) | 1 | 0 | 20 | 24 | 6,7,8,8 | 0.82 (1.48) | 0.89 (1.33) | |||
| 2 | 0 | 20 | 24 | 6,6,8,8 | 0.82 (3.34) | 0.88 (3.17) | ||||
| 3 | +1 | 20 | 24 | 6,7,8,8 | 0.76 (1.16) | 0.69 (0.89) | 20% | 14% | ||
| 4 | +1 | 22 | 26 | 5,6,7,9 | 5,6,8,8 | 1.07 (2.10) | 1.03 (1.70) | |||
| 5 | +2 | 20 | 24 | 4,5,8,8 | 5,6,8,8 | 0.98 (1.39) | 0.99 (0.99) | |||
| (011) | 1 | 0 | 10 | 12 | 7,7,9,9 | 0.78 (1.30) | 0.81 (1.14) | |||
| 2 | 0 | 10 | 12 | 6,6,8,8 | 0.61 (1.32) | 0.68 (1.15) | 22% | 13% | ||
| 3 | +2 | 10 | 12 | 4,4,8,8 | 1.25 (1.68) | 1.35 (1.38) | ||||
| (111) | 1 | 0 | 20 | 24 | 6,7,7,8 | 7,7,8,8 | 1.02 (3.46) | 0.87 (3.29) | ||
| 2 | +1 | 20 | 24 | 5,6,8,8 | 0.83 (1.30) | 0.82 (1.04) | 10% | |||
| 3 | +1 | 22 | 26 | 6,6,7,9 | 1.05 (1.70) | 0.75 (1.11) | 7% | |||
| 4 | +2 | 20 | 24 | 5,5,7,7 | 0.93 (1.22) | 0.95 (1.13) | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anders, J.; Limberg, N.; Paulus, B. First Principle Surface Analysis of YF3 and Isostructural HoF3. Materials 2022, 15, 6048. https://doi.org/10.3390/ma15176048
Anders J, Limberg N, Paulus B. First Principle Surface Analysis of YF3 and Isostructural HoF3. Materials. 2022; 15(17):6048. https://doi.org/10.3390/ma15176048
Chicago/Turabian StyleAnders, Jennifer, Niklas Limberg, and Beate Paulus. 2022. "First Principle Surface Analysis of YF3 and Isostructural HoF3" Materials 15, no. 17: 6048. https://doi.org/10.3390/ma15176048