
Self-Restructuring of Polyhydromethylsiloxanes by the Hydride Transfer Process: A New Approach to the Cross-Linking of Polysiloxanes and to the Fabrication of Thin Polysiloxane Coatings
 ,
,  , , ,
, , ,
Abstract
:1. Introduction

2. Experimental Section
2.1. Materials
2.2. Analytical Methods
2.3. Restructuring of PHMS
2.4. Preparation and Characteristics of Thin Films
2.5. Theoretical Calculations
3. Results and Discussion
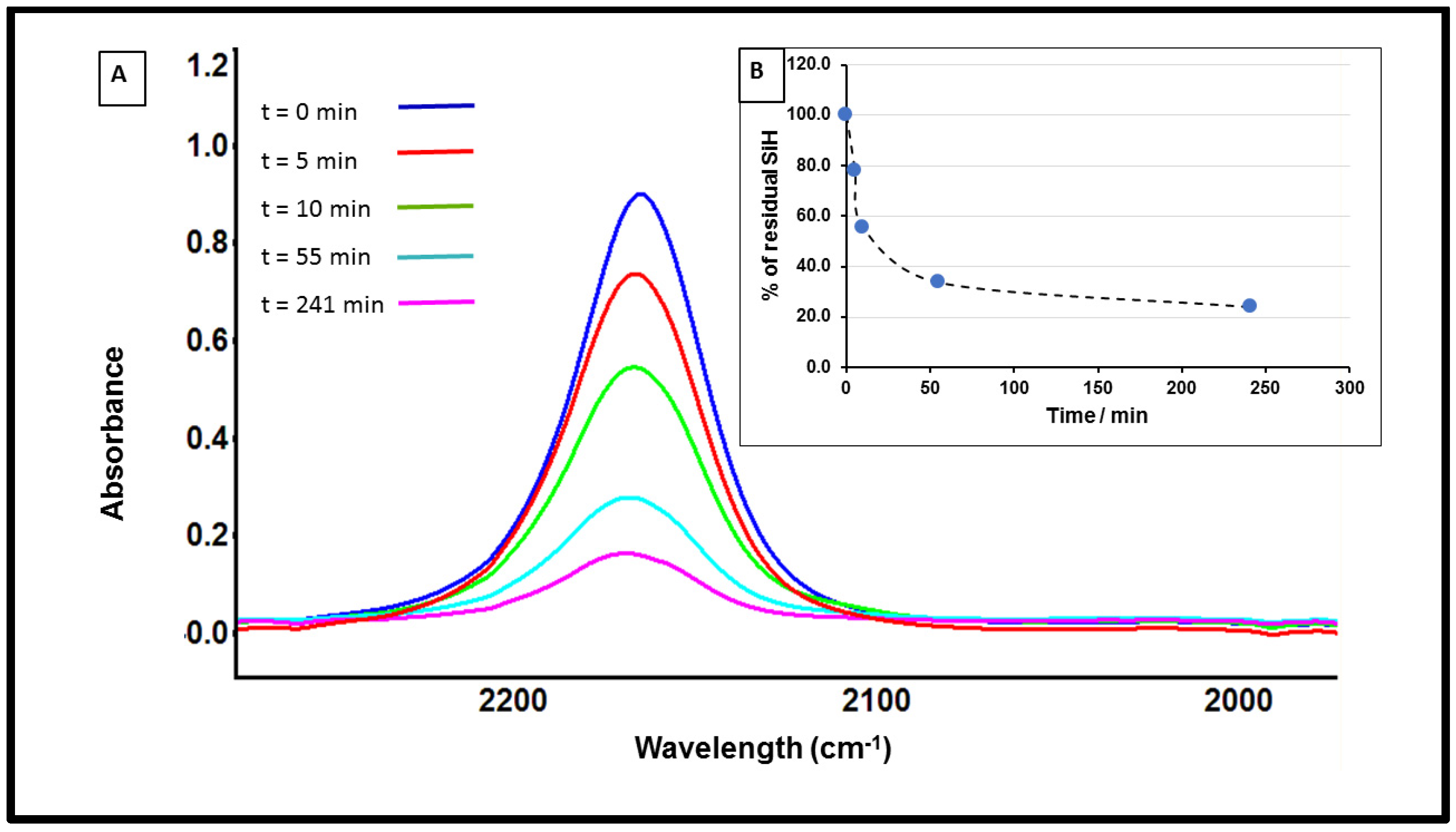
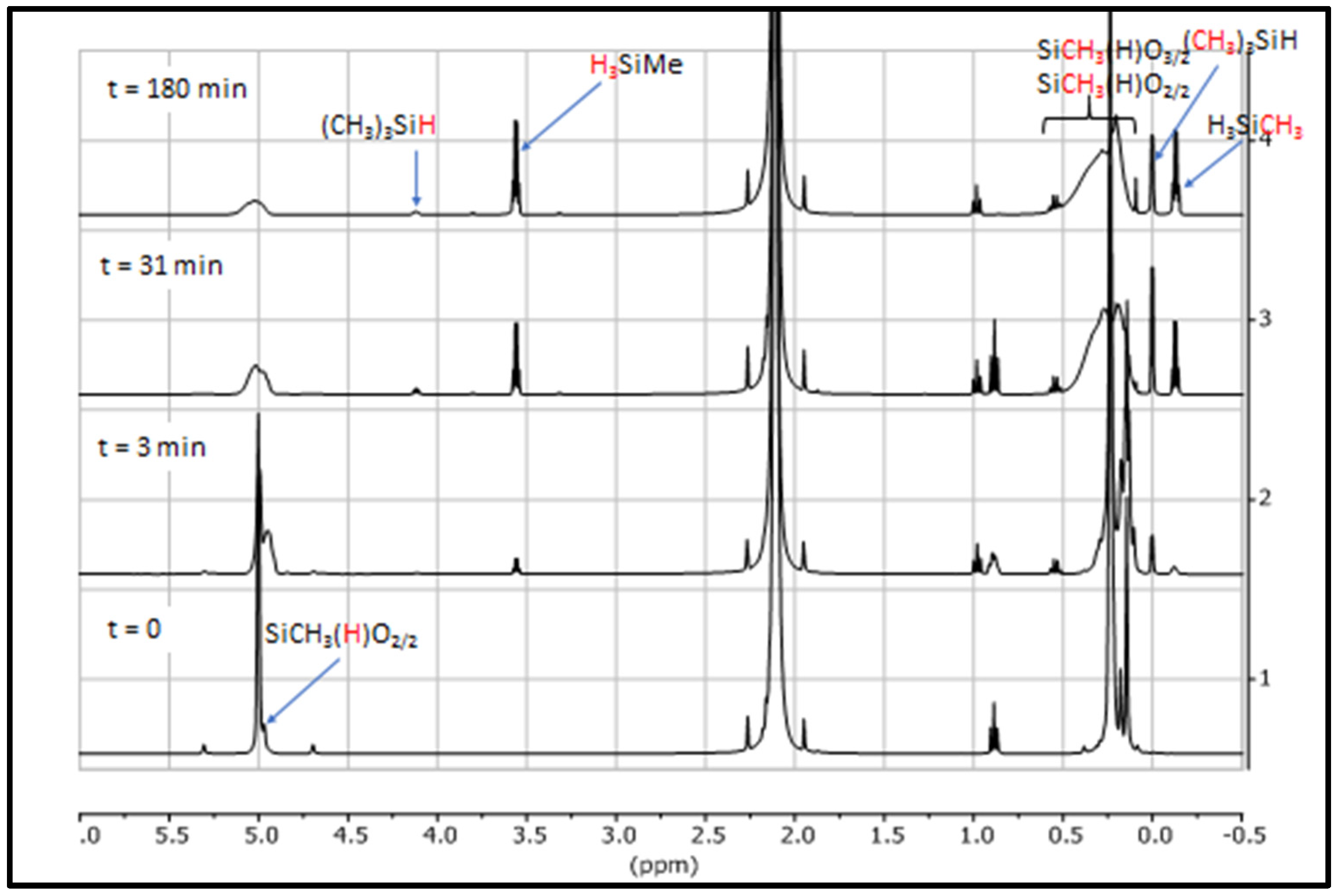
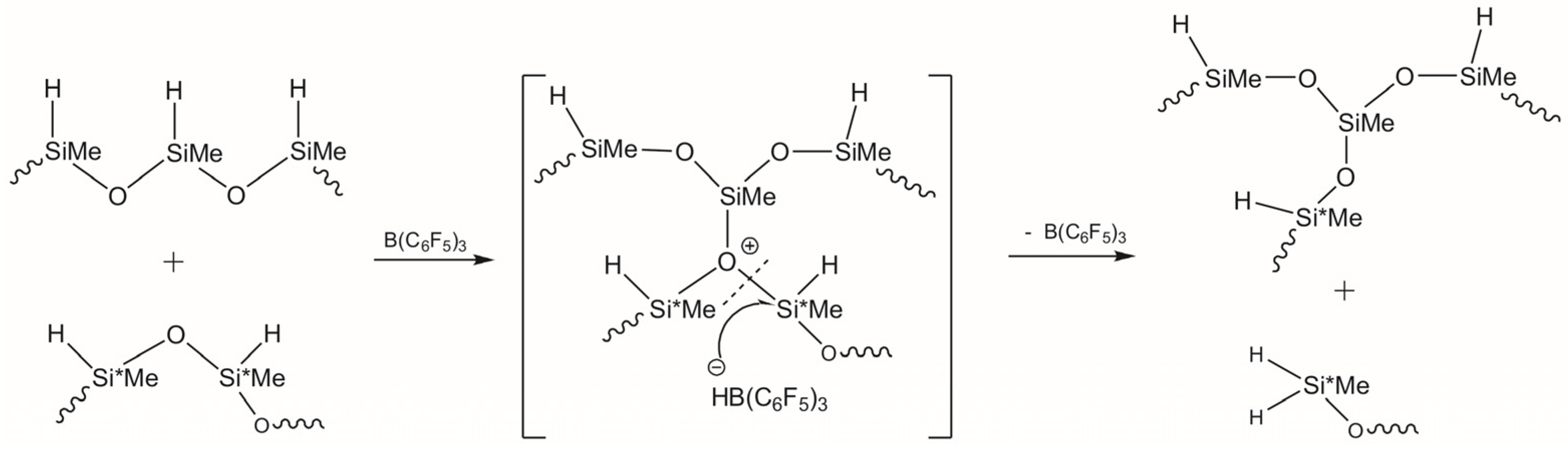
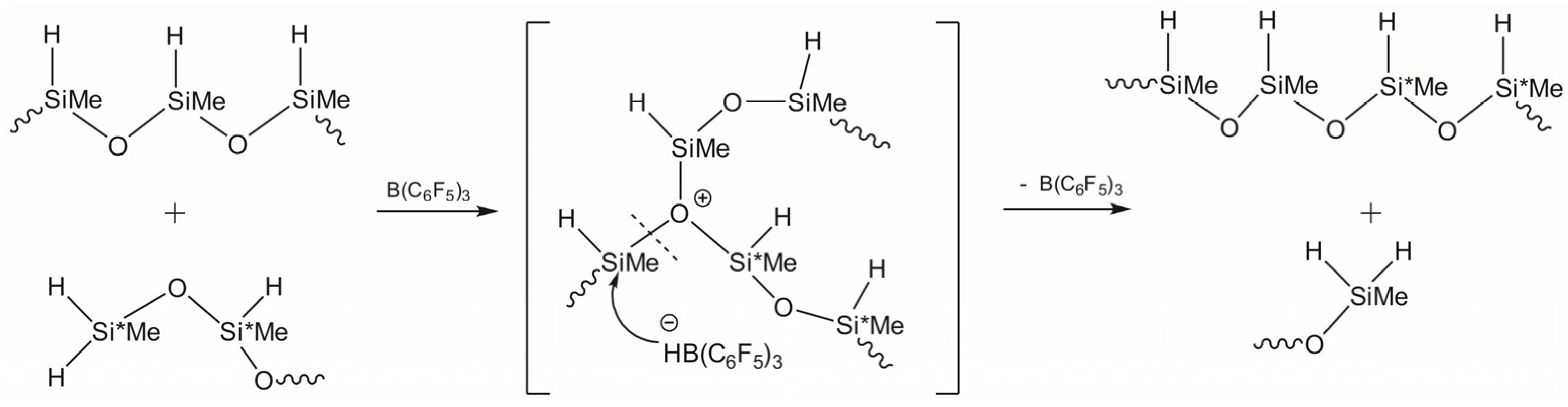
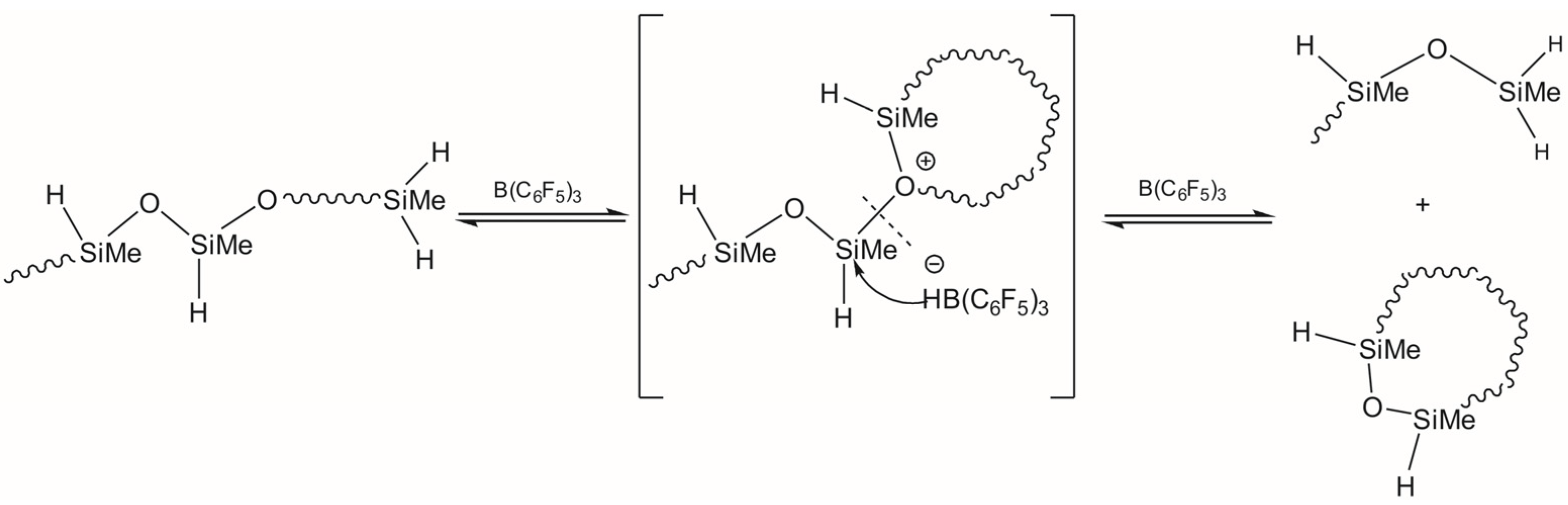
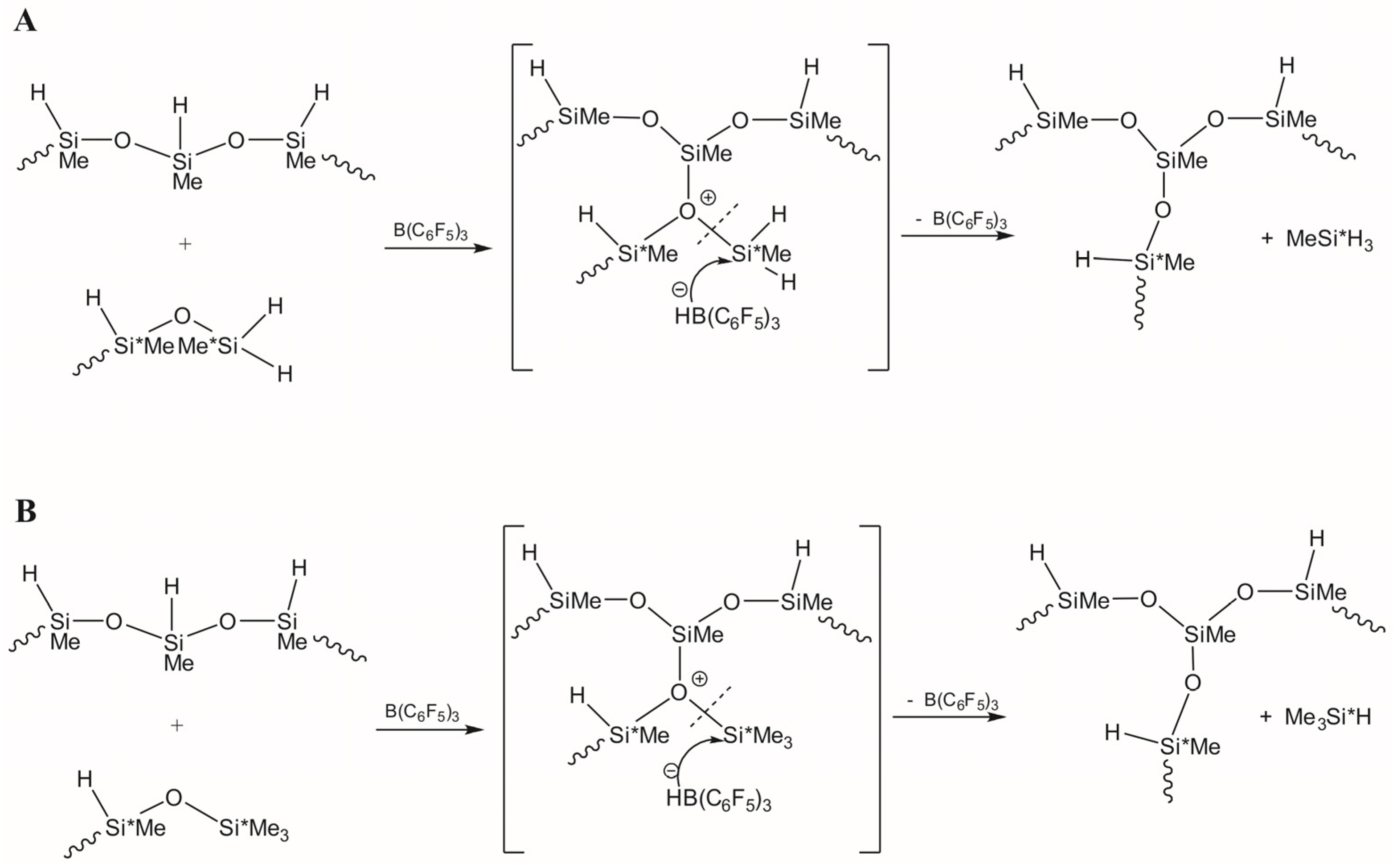
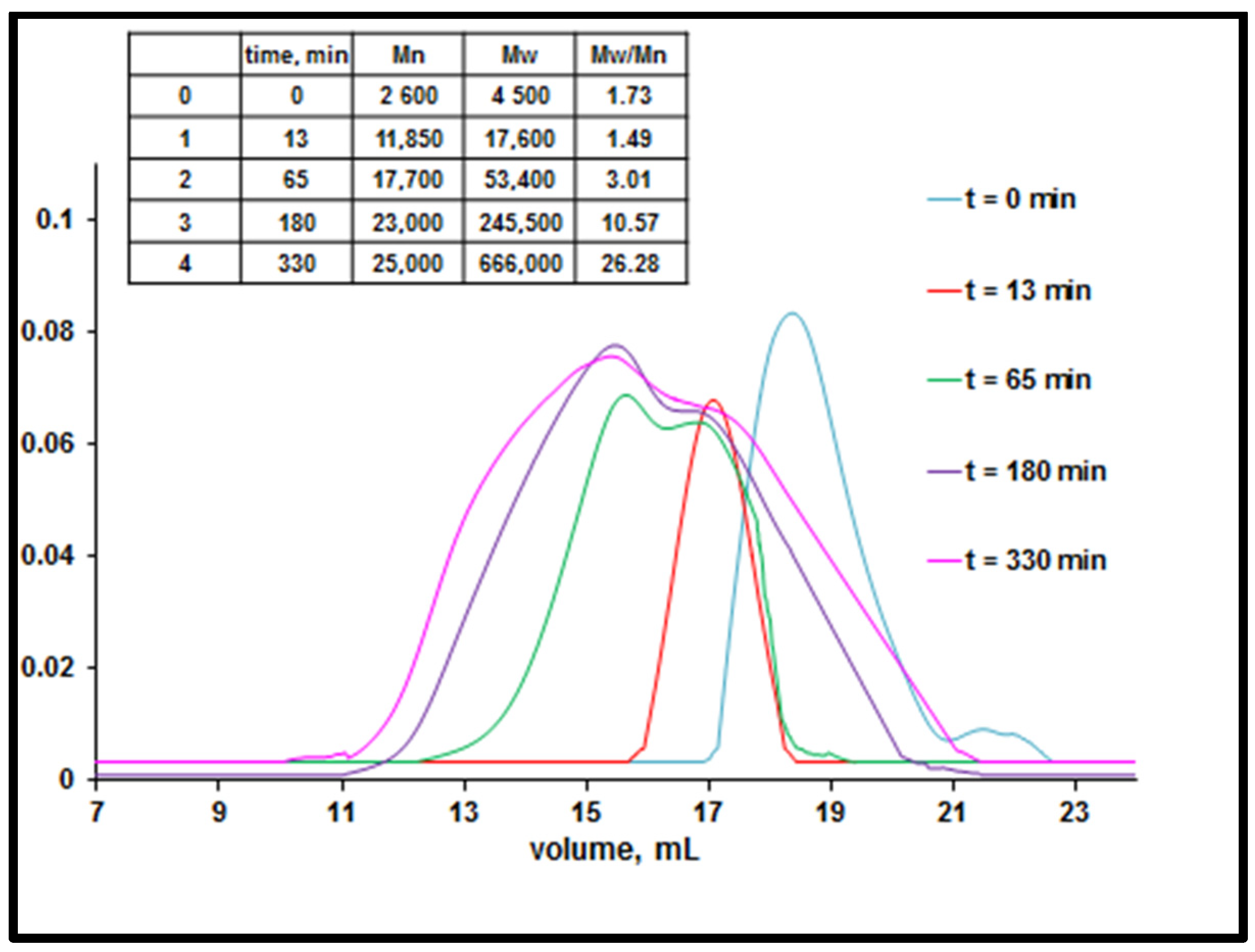
3.1. Study of Restructuring Process
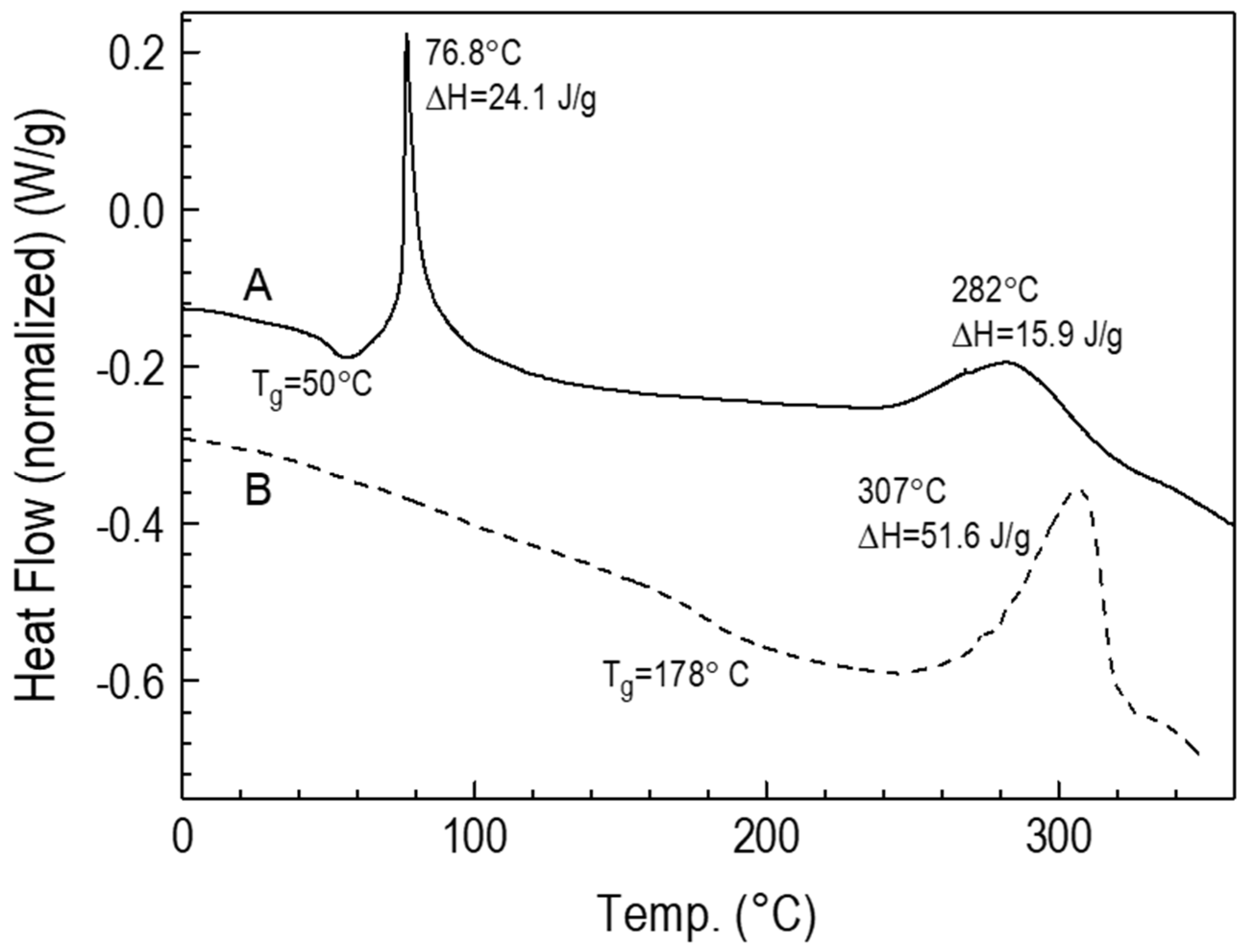
3.2. Preparation and Characteristics of Thin Films
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Noll, W. Chemistry and Technology of Silicones; Academic Press: Cambridge, MA, USA, 1968. [Google Scholar]
- Butts, M.; Cella, J.; Wood, C.D.; Gillette, G.; Kerboua, R.; Leman, J.; Lewis, L.; Rubinsztajn, S.; Schattenmann, F.; Stein, J. Silicones In Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons: Hoboken, NJ, USA, 2000. [Google Scholar] [CrossRef]
- Brook, M.A. Silicon in Organic, Organometallic, and Polymer Chemistry; John Wiley & Sons: New York, NY, USA, 2000. [Google Scholar]
- Tiwari, A.; Soucek, M.D. Concise Encyclopedia of High Performance Silicones; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Wang, D.; Klein, J.; Mejia, E. Catalytic Systems for the cross-linking of organosilicon polymers. Asian J. Chem. 2017, 12, 1180–1197. [Google Scholar] [CrossRef] [PubMed]
- Brinker, C.J.; Scherer, G.W. Sol-gel Science. In The Physics and Chemistry of Sol-Gel Processing; Academic Press: San Diego, CA, USA, 2013. [Google Scholar] [CrossRef]
- Criado, M.; Sobrados, I.; Sanz, J. Polysiloxane hybrids via Sol-gel process: Effect of temperature on network formation. Coatings 2020, 10, 677. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Z.; Zhu, T.; Li, Z.; Wang, J.; Cheng, Y. Multi-benzocyclobutene functionalized siloxane monomers prepared by Piers-Rubinsztajn reaction for low-k materials. Eur. Polym. J. 2020, 126, 109562. [Google Scholar] [CrossRef]
- Robeyns, C.; Picard, L.; Ganachaud, F. Synthesis, characterization and modification of silicone resins: An “Augmented Review”. Prog. Org. Coat. 2018, 125, 287–315. [Google Scholar] [CrossRef]
- Dascalu, M.; Dünki, S.J.; Quinsaat, J.-E.Q.; Ko, Y.S.; Opris, D.M. Synthesis of silicone elastomers containing trifluoropropyl groups and their use in dielectric elastomer transducers. RSC Adv. 2015, 5, 104516–104523. [Google Scholar] [CrossRef]
- Heiner, J.; Sternberg, B.; Persson, M. Crosslinking of siloxane elastomers. Polym. Test. 2003, 22, 253–257. [Google Scholar] [CrossRef]
- Yuan, J.; Huang, Y.; Li, H.; Jiang, L.; Dan, Y. Preparation and anti-icing performance of cross-linked polysiloxane coating silicone oil. React. Funct. Polym. 2022, 170, 105124. [Google Scholar] [CrossRef]
- Shi, Y.; Cai, J.; Wu, X.; Cheng, Y. Benzocyclobutene-functionalized hyperbranched polysiloxane for low-k materials with good thermostability. Des. Monomers Polym. 2021, 4, 285–292. [Google Scholar] [CrossRef]
- Mozelewska, K.; Antosik, A.K. Influence of silicone additives on the properties of pressure-sensitive adhesives. Materials 2022, 15, 5713. [Google Scholar] [CrossRef]
- Wang, G.; Wang, J.; Park, C.B.; Kim, Y.-W. Cross-linking behavior of polysiloxane in preceramic foam processing. J. Mater. Sci. 2004, 39, 4913–4915. [Google Scholar] [CrossRef]
- Chojnowski, J.; Slomkowski, S.; Fortuniak, W.; Mizerska, U.; Pospiech, P. Hydrophilic polysiloxane microspheres and ceramic SiOC microspheres derived from them. J. Inorg. Organomet. Polym. Mater. 2020, 30, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.-F.J.; Chen, J.-B. Study on the synthesis and application of silicone resin containing phenyl group. J. Sol-Gel Sci. Technol. 2015, 76, 66–73. [Google Scholar] [CrossRef]
- Marciniec, B. Hydrosilylation. A Comprehensive Review on Recent Advances; Marciniec, B., Ed.; Springer: Dordrecht, The Netherlands, 2009; Volume 1, pp. 159–189. [Google Scholar]
- Kownacki, I.; Marciniec, B.; Macina, A.; Rubinsztajn, S.; Lamb, D. Catalytic activity of iridium siloxide complexes in cross-linking of silicones by hydrosilylation. Appl. Catal. A-Gen. 2007, 317, 53–57. [Google Scholar] [CrossRef]
- Dobrynin, M.V.; Pretorius, C.; Kama, D.V.; Roodt, A.; Boyarsky, V.P.; Islamova, R.M. Rhodium I-catalyzed cross-linking of polysiloxanes conducted at room temperature. J. Catal. 2019, 372, 193–200. [Google Scholar] [CrossRef]
- Islamova, R.M.; Dobrynin, M.V.; Ivanov, D.M.; Vlasov, A.V.; Kaganova, E.V.; Grigoryan, G.V.; Kukushkin, V.Y. Bis-nitrile and bis-dialkylcyanamide platinum(II) complexes as efficient catalysts for hydrosilylation cross-linking of siloxane polymers. Molecules 2016, 12, 311. [Google Scholar] [CrossRef] [Green Version]
- Foran, G.Y.; Harris, K.J.; Brook, M.A.; Macphail, B.; Goward, G.R. Solid State NMR Study of Boron Coordination Environments in Silicone Boronate (SiBA) Polymers. Macromolecules 2019, 52, 1055–1064. [Google Scholar] [CrossRef]
- Kim, E.E.; Kononevich, Y.U.N.; Anisimov, A.A.; Buzin, M.I.; Vasilev, V.G.; Korlyukov, A.A.; Ionov, D.S.; Khanin, D.A.; Shtykova, E.V.; Volkov, V.V.; et al. Cross-linked polymer networks based on polysiloxane and nickel β-diketonate precursors. React. Func. Polym. 2021, 164, 104896. [Google Scholar] [CrossRef]
- Deriabin, K.V.; Yaremenko, I.A.; Chislov, M.V.; Fleury, F.; Terentev, A.O.; Islamova, R.M. Similar nature leads to improved properties: Cyclic organosilicon triperoxides as promising curing agents for liquid polysiloxanes. New J. Chem. 2018, 42, 15006. [Google Scholar] [CrossRef]
- Rubinsztajn, S.; Chojnowski, J.; Cypryk, M.; Mizerska, U.; Fortuniak, W.; Bak-Sypien, I.I. Kinetic and mechanistic studies of the transformation of the catalyst, tris (pentafluorophenyl) borane, in the presence of silyl and germyl hydrides. J. Catal. 2019, 379, 90–99. [Google Scholar] [CrossRef]
- Chojnowski, J.; Kurjata, J.; Fortuniak, W.; Rubinsztajn, S.; Trzebicka, B. Hydride Transfer Ring-Opening Polymerization of a Cyclic Oligomethylhydrosiloxane. Route to a Polymer of Closed Multicyclic Structure. Macromolecules 2012, 45, 2654–2661. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. B.01, Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.L.; Zheng, J.; Alecu, I.M.; Lynch, B.J.; Zhao, Y.; Truhlar, D.G. Database of Fequency Scale Factors for Electronic Model Chemistries. Available online: https://comp.chem.umn.edu/freqscale/version3b2.htm (accessed on 1 January 2022).
- Zhang, Y.; Caporaso, L.; Cavallo, L.; Chen, E.Y.-X. Hydride-Shuttling Chain-Transfer Polymerization of Methacrylates Catalyzed by Metallocenium Enolate Metallacycle−Hydridoborate Ion Pairs. J. Am. Chem. Soc. 2011, 133, 1572–1588. [Google Scholar] [CrossRef] [PubMed]
- Houghton, A.; Hurmalainen, J.; Mansikkamaki, A.; Piers, W.; Tuononen, H. Direct observation of a borane-silane complex involved in frustrated Lewis-pair-mediated hydrosilylations. Nat. Chem. 2014, 6, 983–988. [Google Scholar] [CrossRef]
- Chojnowski, J.; Rubinsztajn, S.; Cella, J.; Fortuniak, W.; Cypryk, M.; Kurjata, J.; Kazmierski, K. Mechanism of the B(C6F5)(3)-catalyzed reaction of silyl hydrides with alkoxysilanes. Kinetic and spectroscopic studies. Organometallics 2005, 24, 6077–6084. [Google Scholar] [CrossRef]
- Liao, M.; Schneider, A.F.; Laengert, S.E.; Gale, C.B.; Chen, Y.; Brook, M.A. Living synthesis of silicone polymers controlled by humidity. Eur. Polym. J. 2018, 107, 287–293. [Google Scholar] [CrossRef]
- Schneider, A.F.; Laidley, E.; Brook, M.A. Facile Synthesis of Cx (AB) yCx Triblock Silicone Copolymers Utilizing Moisture Mediated Living-End Chain Extension. Macromol. Chem. Phys. 2019, 220, 1800575. [Google Scholar] [CrossRef]
- Yousefi, A.; Lafleur, P.; Gauvin, R. Kinetic studies of thermoset cure reactions: A review. Polym. Compos. 1997, 18, 157–168. [Google Scholar] [CrossRef]
- Zhang, Y.; Adams, R.; da Silva, L.F. Effects of curing cycle and thermal history on the glass transition temperature of adhesives. J. Adhes. 2014, 90, 327–345. [Google Scholar] [CrossRef]
- Bahloul-Hourlier, D.; Latournerie, J.; Dempsey, P. Reaction pathways during the thermal conversion of polysiloxane precursors into oxycarbide ceramics. J. Eur. Ceram. Soc. 2005, 25, 979–985. [Google Scholar] [CrossRef]
- Wong, M.Y.; Schneider, A.F.; Lu, G.; Chen, Y.; Brook, M.A. Autoxidation: Catalyst-free route to silicone rubbers by crosslinking Si–H functional groups. Green Chem. 2019, 21, 6483–6490. [Google Scholar] [CrossRef]


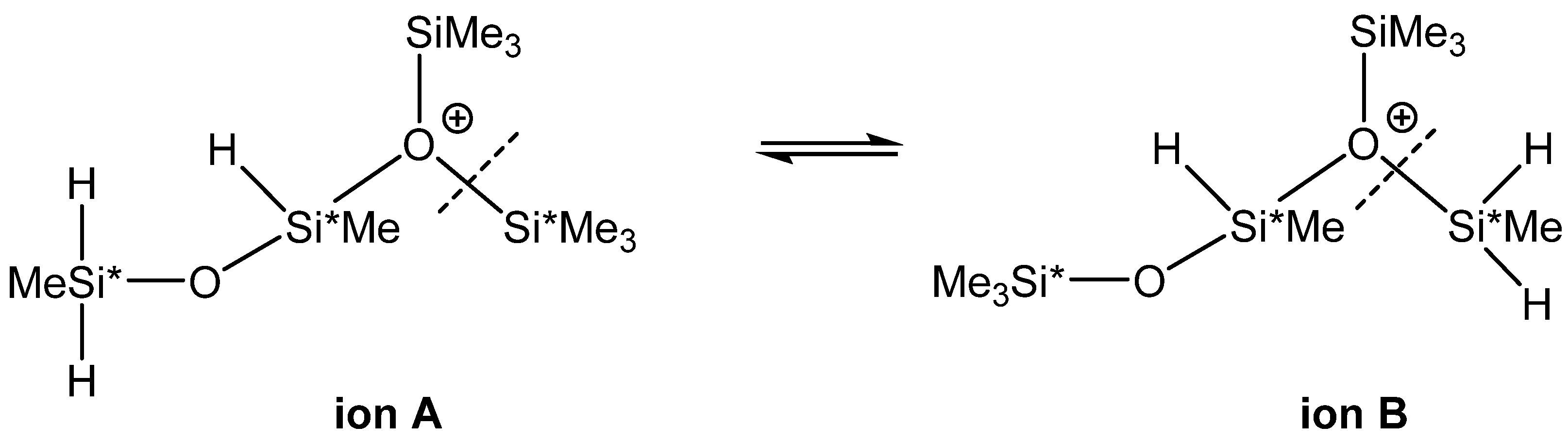



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
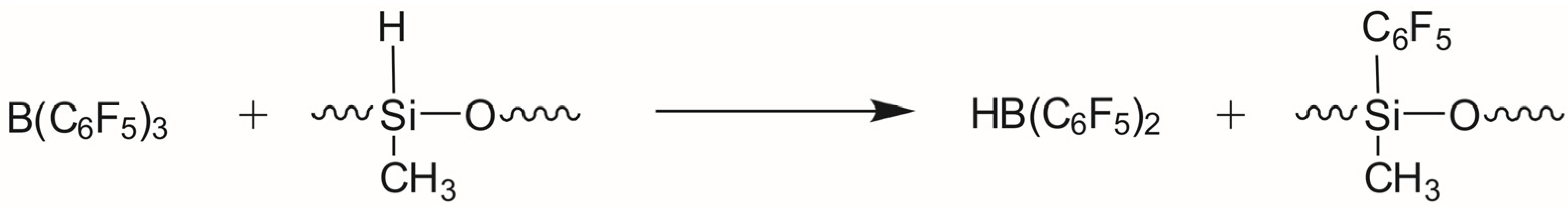
| No. | Reaction | ΔH298 Kcal/mol | ΔG298 Kcal/mol | r(H-B) Å |
|---|---|---|---|---|
| 1 | 2 Me3SiOSiHMeOSiMe3 → Me3SiOSiMeH2 + (Me3SiO)3SiMe | −3.2 | −5.3 | |
| 2 | Me3SiOSiHMeOSiMe3 + Me3SiOSiMeH2 → MeSiH3 + (Me3SiO)3SiMe | −10.1 | −7.4 | |
| 3 | Me3SiH + B(C6F5)3 → Me3SiH-B(C6F5)3 | −8.5 | 4.0 | 1.516 |
| 4 | Me2SiH2 + B(C6F5)3 → Me2SiH2-B(C6F5)3 | −5.6 | 4.9 | 2.302 |
| 5 | MeSiH3 + B(C6F5)3 → MeSiH3-B(C6F5)3 | −3.9 | 5.7 | 2.540 |
| 6 | Me3SiOSiHMeOSiMe3 + B(C6F5)3 → (Me3SiO)2SiMeH-B(C6F5)3 | −11.3 | 4.7 | 1.525 |
| 7 | (Me3Si)2O(+)(SiHMeOSiMeH2) (A) → (Me3Si)(MeH2Si)O(+)(SiHMeOSiMe3) (B) | −2.4 | −3.2 |
| Experiment Number | Initial PHMS Concentration w% | [DH] mol/L | Time to Gelation Min | DH Conversion at Gelation % |
|---|---|---|---|---|
| 1 | 10 | 1.3 | No Gel | No Gel |
| 2 | 20 | 2.6 | 260 | 76 |
| 3 | 30 | 4.0 | 6 | 60 |
| Film # | Polymer Concentration/wt% | Time of Reaction/h | Initial % HMeSiO Conversion | Film Quality | % HMeSiO Conversion after 24 h @ RT | % HMeSiO Conversion after Postbake @150 °C |
|---|---|---|---|---|---|---|
| 1 | 10 | 0.67 | 56.7 | Poor | 71.9 | 83.7 |
| 2 | 10 | 3 | 72.6 | Good | 77.4 | 88.5 |
| 3 | 10 | 24 | 82 | Good | ND | 86 |
| 4 | 20 | 2 | 70.4 | Good | 77 | 87.8 |
| 5 | 20 | 8 | 77 | Gel/Poor | ND | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizerska, U.; Rubinsztajn, S.; Chojnowski, J.; Cypryk, M.; Uznanski, P.; Walkiewicz-Pietrzykowska, A.; Fortuniak, W. Self-Restructuring of Polyhydromethylsiloxanes by the Hydride Transfer Process: A New Approach to the Cross-Linking of Polysiloxanes and to the Fabrication of Thin Polysiloxane Coatings. Materials 2022, 15, 6981. https://doi.org/10.3390/ma15196981
Mizerska U, Rubinsztajn S, Chojnowski J, Cypryk M, Uznanski P, Walkiewicz-Pietrzykowska A, Fortuniak W. Self-Restructuring of Polyhydromethylsiloxanes by the Hydride Transfer Process: A New Approach to the Cross-Linking of Polysiloxanes and to the Fabrication of Thin Polysiloxane Coatings. Materials. 2022; 15(19):6981. https://doi.org/10.3390/ma15196981
Chicago/Turabian StyleMizerska, Urszula, Slawomir Rubinsztajn, Julian Chojnowski, Marek Cypryk, Pawel Uznanski, Agnieszka Walkiewicz-Pietrzykowska, and Witold Fortuniak. 2022. "Self-Restructuring of Polyhydromethylsiloxanes by the Hydride Transfer Process: A New Approach to the Cross-Linking of Polysiloxanes and to the Fabrication of Thin Polysiloxane Coatings" Materials 15, no. 19: 6981. https://doi.org/10.3390/ma15196981