New Insights on the Electronic-Structural Interplay in LaPdSb and CePdSb Intermetallic Compounds


 ,
,
Abstract
:1. Introduction
2. Methods
3. Experimental Results: Neutron and X-ray Diffraction—Diffuse Scattering

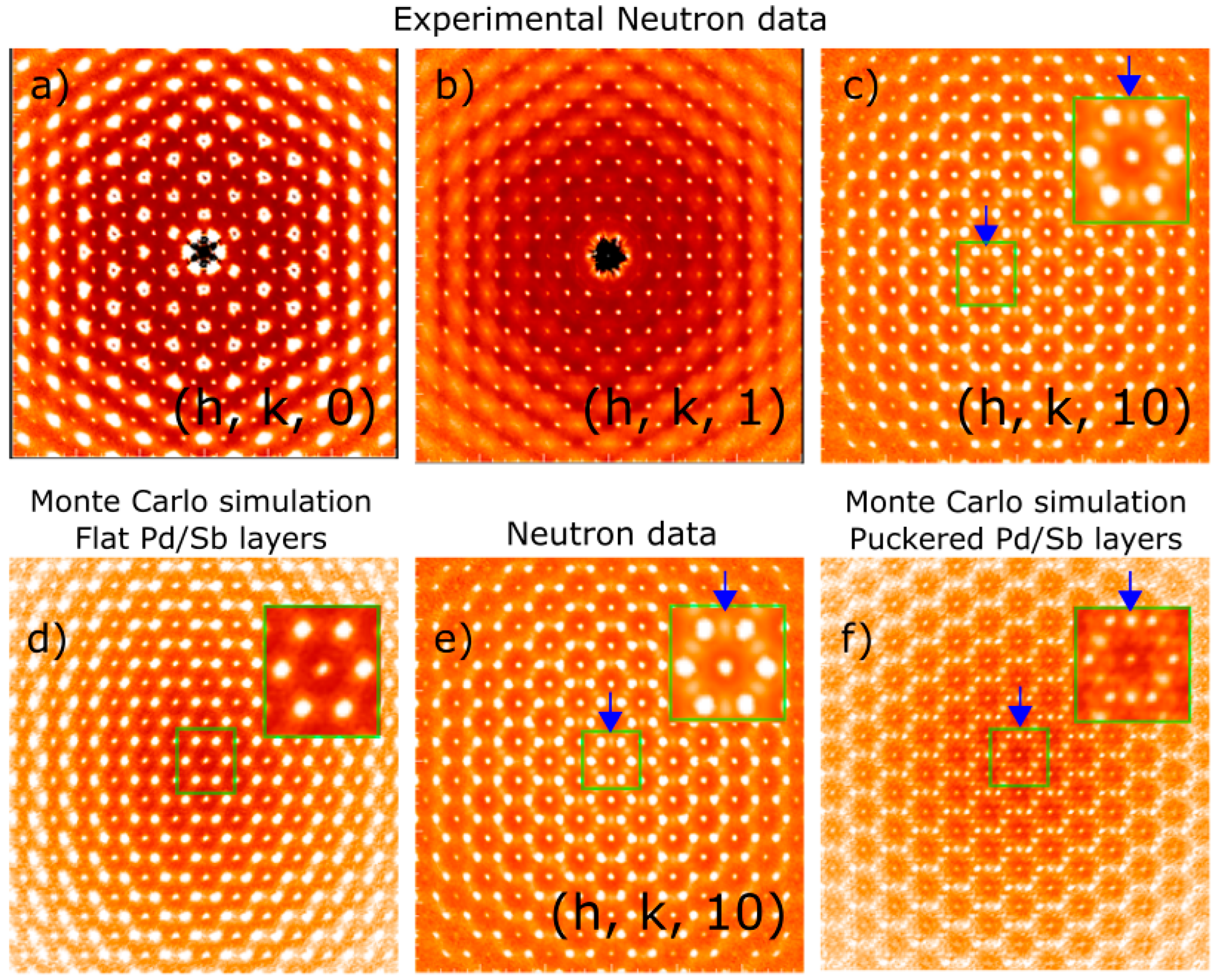

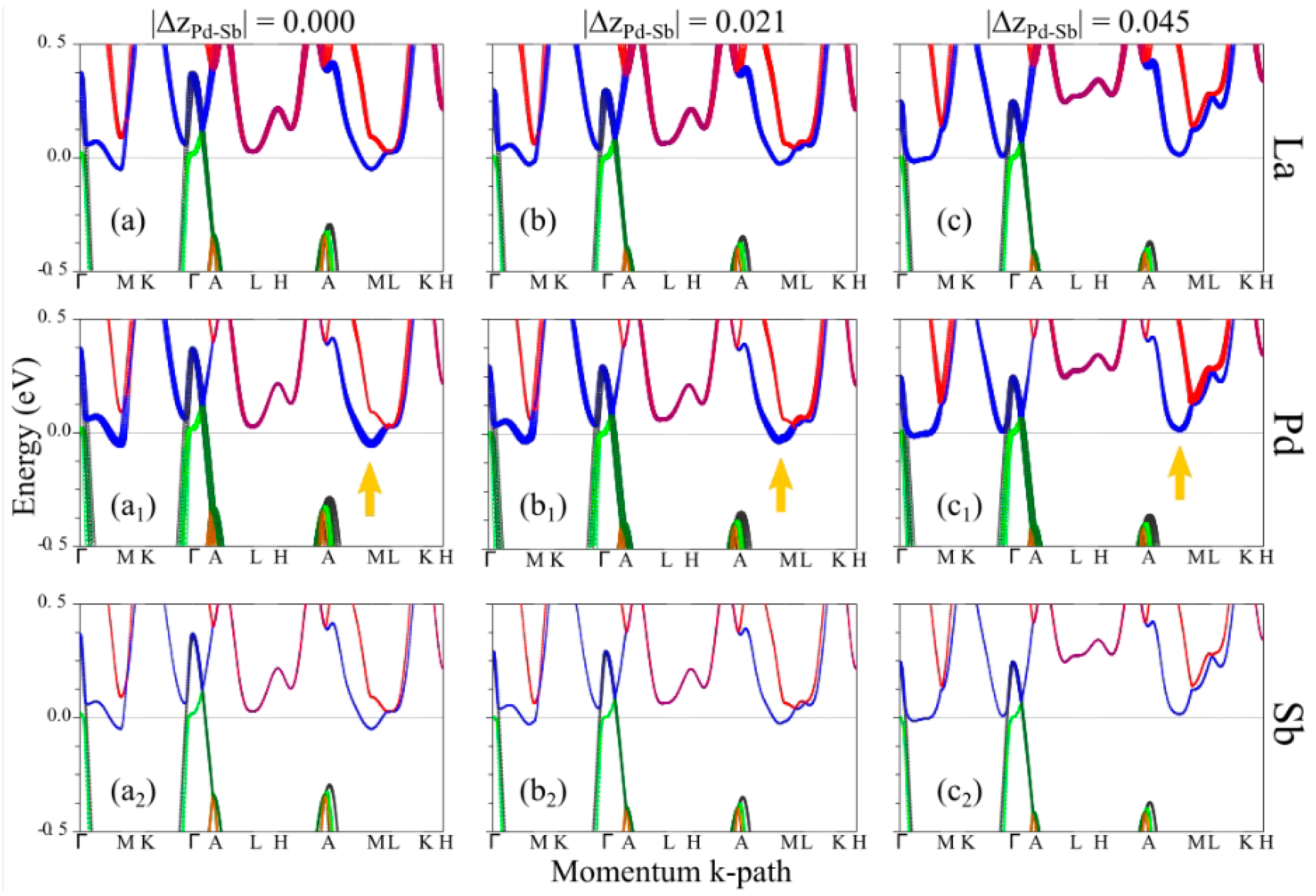
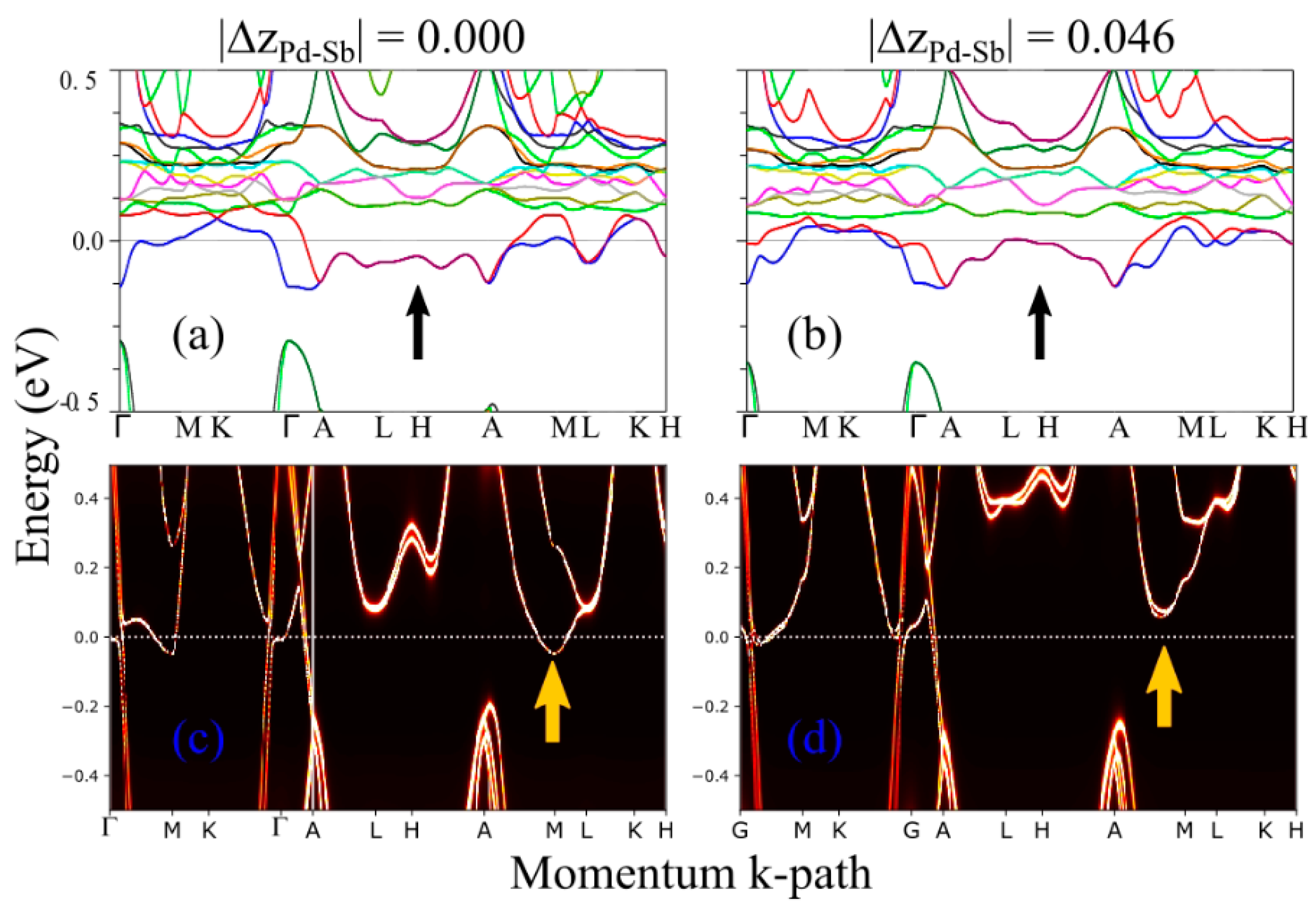
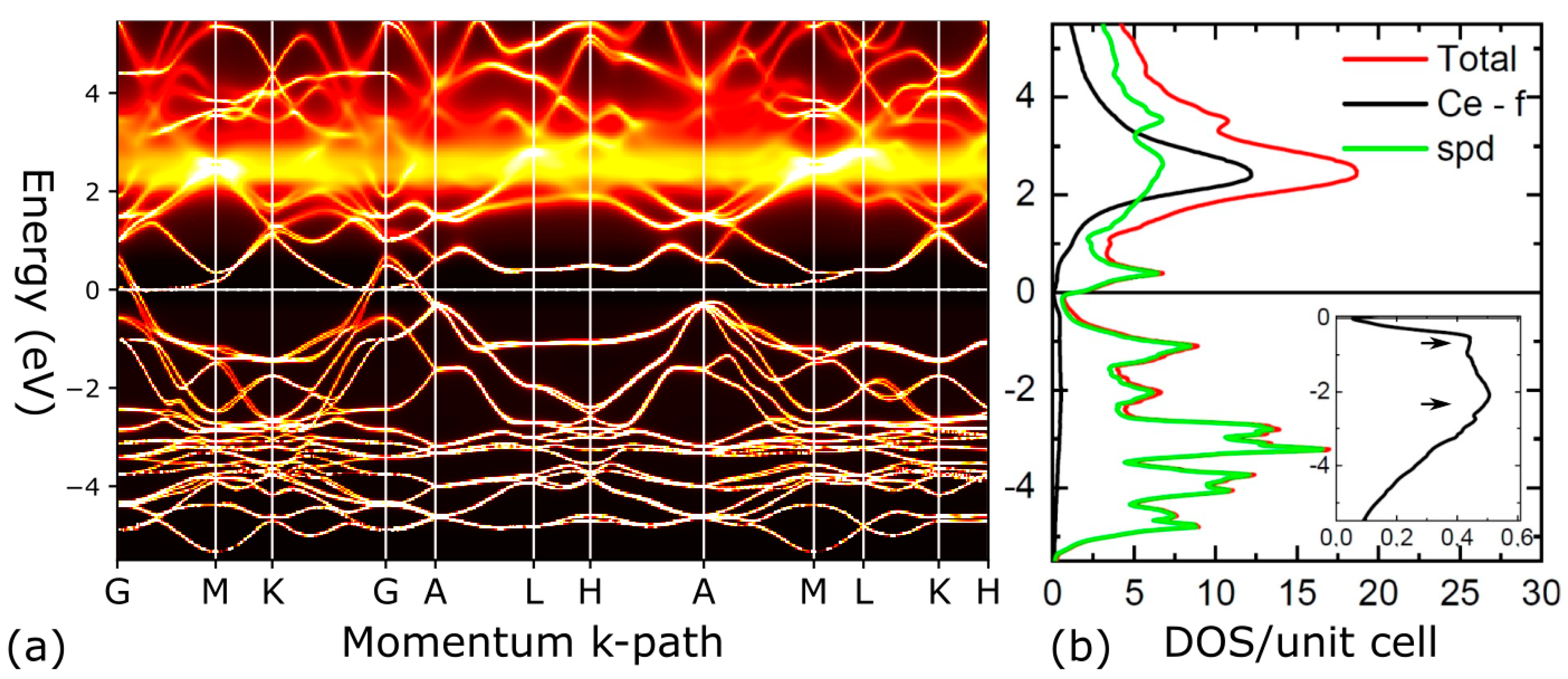
4. Theoretical Results: Microscopic Origin of the Puckering Distortion
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malik, S.K.; Adroja, D.T. CePdSb: A possible ferro magnetic Kondo-lattice system. Phys. Rev. B 1991, 43, 6295. [Google Scholar] [CrossRef] [PubMed]
- Riedi, P.C.; Armitage, J.G.N.; Lord, J.S.; Adroja, D.T.; Rainford, B.D.; Fort, D. A ferromagnetic Kondo compound: CePdSb. Phys. B 1994, 199, 558–560. [Google Scholar] [CrossRef]
- Zygmunt, A.; Szytula, A. Magnetic properties of RPdSn and RPdSb compounds. J. Alloy. Compd. 1995, 219, 185–188. [Google Scholar] [CrossRef]
- Katoh, K.; Ochiai, A.; Suzuki, T. Magnetic and Transport properties of CePdAs and CePdSb. Phys. B 1996, 223, 340–343. [Google Scholar] [CrossRef]
- Katoh, K.; Takabatake, T.; Ochiai, A.; Uesawa, A.; Suzuki, T. Quasi-two-dimensional conductivity in CePdSb an CePdSb. Phys. B 1997, 230, 159–161. [Google Scholar] [CrossRef]
- Katoh, K.; Takabatake, T.; Oguro, I.; Ochiai, A.; Uesawa, A.; Suzuki, T. Anisotropic Behavior of Magnetic and transport Properties I CePdSb and CePtSb. J. Phys. Soc. Jpn. 1999, 68, 613–619. [Google Scholar] [CrossRef]
- Lord, J.S.; Tomka, G.J.; Riedi, P.C.; Thornton, M.J.; Rainford, B.D.; Adroja, D.T.; Fort, D. A nuclear magnetic resonance investigation of the ferromagnetic phase of CePdSb as a function of temperature and pressure. J. Phys. Condens. Matter 1996, 8, 5475–5482. [Google Scholar] [CrossRef]
- Ślebarski, A. Half-metallic ferromagnetic ground state in CePdSb. J. Alloy. Compd. 2006, 423, 15–20. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Mohapatra, N. Electrical transport properties of ternary half-Heusler LaPdSb. AIP Conf. Proc. 2018, 1942, 110014. [Google Scholar]
- Ślebarski, A.; Głogowski, W.; Jezierski, A.; Deniszczyk, J.; Czopnik, A.; Zygmunt, A. Electronic structure and magnetic properties of CePdSb and Ce1−xLaxPdSb. Phys. Rev. B 2004, 70, 184429. [Google Scholar] [CrossRef]
- Iwasaki, T.; Suga, S.; Imada, S.; Kuwata, Y.; Muro, T.; Ueda, S.; Harada, H.; Tsunekawa, M.; Matsushita, T.; Sekiyama, A.; et al. High resolution resonance photoemission, XPS and inverse photoemission spectroscopy of CePdX (X = As, Sb). J. Electron Spectrosc. Relat. Phenom. 1998, 88, 309–314. [Google Scholar] [CrossRef]
- Iwasaki, T.; Suga, S.; Imada, S.; Sekiyama, A.; Matsuda, K.; Kotsugi, M.; An, K.-S.; Muro, T.; Ueda, S.; Matsushita, T.; et al. Bulk and surface electronic structures of CePdX (X = As, Sb) studied by 3d–4f resonance photoemission. Phys. Rev. B 2000, 61, 4621. [Google Scholar] [CrossRef]
- Sekimoto, T.; Kurosaki, K.; Muta, H.; Yamanaka, S. LnPdSb (Ln = La, Gd) (Ln = La, Gd): Promising intermetallics with large carrier mobility for high performance p-type thermoelectric materials. Appl. Phys. Lett. 2006, 89, 092108. [Google Scholar] [CrossRef]
- Sekimoto, T.; Kurosaki, K.; Muta, H.; Yamanaka, S. High Temperature Thermoelectric Properties of LnPdX (Ln = lanthanide; X = Sb, Bi) Ternary Compounds. MRS Online Proc. Libr. 2005, 886, 804. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.A.; Lakshminarasimhan, N.; Mohapatra, N. Multi-functional properties of non-centrosymmetric ternary half-Heuslers, RPdSb (R = Er and Ho). J. Phys. D Appl. Phys. 2018, 51, 265004. [Google Scholar] [CrossRef]
- Malik, S.K.; Adroja, D.T. Magnetic behaviour of RPdSb (R = rare earth) compounds. J. Magn. Magn. Mater. 1991, 102, 42–46. [Google Scholar] [CrossRef]
- Rainford, B.D.; Adroja, D.T.; Neville, A.; Fort, D. Spin dynamics and crystal fields of CePdSb. Phys. B Condens. Matter 1995, 206, 209–211. [Google Scholar] [CrossRef]
- Keen, D.A.; Gutmann, M.J.; Wilson, C.C. SXD—The single-crystal diffractometer at the ISIS spallation neutron source. J. Appl. Cryst. 2006, 39, 714. [Google Scholar] [CrossRef]
- Gutmann, M.J. SXD2001; ISIS Facility, Rutherford Appleton Laboratory: Oxfordshire, UK, 2005. [Google Scholar]
- Petricek, V.; Dusek, M.; Palatinus, L. Crystallographic Computing System JANA2006: General Features. Z. Kristallogr. 2014, 229, 345–352. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Cryst. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Koch, R.J.; Roth, N.; Liu, Y.; Ivashko, O.; Dippel, A.-C.; Petrovic, C.; Iversen, B.B.; Zimmermann, M.v.; Bozin, E.S. On single-crystal total scattering data reduction and correction protocols for analysis in direct space. Acta Cryst. 2021, 77, 611–636. [Google Scholar]
- Goossens, D.J. Monte Carlo Modelling of Single-Crystal Diffuse Scattering from Intermetallics. Metals 2016, 6, 33. [Google Scholar] [CrossRef]
- Gutmann, M.J. Accelerated computation of diffuse scattering patterns and application to magnetic neutron scattering. J. Appl. Cryst. 2010, 43, 250–255. [Google Scholar] [CrossRef]
- Blaha, P.; Schwarz, K.; Tran, F.; Laskowski, R.; Madsen, G.K.H.; Marks, L.D. WIEN2k: An APW + lo program for calculating the properties of solids. J. Chem. Phys. 2020, 152, 074101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Marks, L.D. Fixed-Point Optimization of Atoms and Density in DFT. J. Chem. Theory Comput. 2013, 9, 2786–2800. [Google Scholar] [CrossRef]
- Haule, K.; Pascut, G.L. Forces for structural optimizations in correlated materials within a DFT + embedded DMFT functional approach. Phys. Rev. B 2016, 94, 195146. [Google Scholar] [CrossRef] [Green Version]
- Haule, K.; Yee, C.-H.; Kim, K. Dynamical mean-field theory within the full-potential methods: Electronic structure of CeIrIn5, CeCoIn5, and CeRhIn5. Phys. Rev. B 2010, 81, 195107. [Google Scholar] [CrossRef] [Green Version]
- Haule, K. Structural predictions for correlated electron materials using the functional dynamical mean field theory approach. J. Phys. Soc. Jpn. 2018, 87, 041005. [Google Scholar] [CrossRef]
- Paul, A.; Birol, T. Applications of DFT + DMFT in materials science. Annu. Rev. Mater. Res. 2019, 49, 31. [Google Scholar] [CrossRef] [Green Version]
- Kotliar, G.; Savrasov, S.Y.; Haule, K.; Oudovenko, V.S.; Parcollet, O.; Marianetti, C.A. Electronic structure calculations with dynamical mean-field theory. Rev. Mod. Phys. 2006, 78, 865. [Google Scholar] [CrossRef] [Green Version]
- Georges, A.; Kotliar, G. Hubbard model in infinite dimensions. Phys. Rev. B 1992, 45, 6479–6483. [Google Scholar] [CrossRef] [PubMed]
- Vollhardt, D. Dynamical mean-field theory for correlated electrons. Ann. Der. Physik. 2012, 524, 1–19. [Google Scholar] [CrossRef]
- Georges, A. Strongly Correlated Electron Materials: Dynamical Mean-Field Theory and Electronic Structure. arXiv 2004, arXiv:cond-mat/0403123. [Google Scholar]
- Haule, K. Exact Double Counting in Combining the Dynamical Mean Field Theory and the Density Functional Theory. Phys. Rev. Lett. 2015, 115, 196403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haule, K. Quantum Monte Carlo impurity solver for cluster dynamical mean-field theory and electronic structure calculations with adjustable cluster base. Phys. Rev. B 2007, 75, 155113. [Google Scholar] [CrossRef] [Green Version]
- Werner, P.; Comanac, A.; Medici, L.D.; Troyer, M.; Millis, A.J. Continuous-time solver for quantum impurity models. Phys. Rev. Lett. 2006, 97, 076405. [Google Scholar] [CrossRef] [Green Version]
- Haule, K. Analytical Continuation: DFT + Embedded DMFT Functional. Unpublished. Available online: https://hauleweb.rutgers.edu/tutorials/(accessed on 20 October 2022).
- Schwarz, K.; Blaha, P.; Trickey, S. Electronic structure of solids with WIEN2k. Mol. Phys. 2010, 108, 3147–3166. [Google Scholar] [CrossRef]
- Pascut, G.L.; Widom, M.; Haule, K.; Quader, K.F. First-principles study of the electronic structure and the Fermi surface in rare-earth filled skutterudites RPt4Ge12. Phys. Rev. B 2019, 100, 125114. [Google Scholar] [CrossRef] [Green Version]
- Lanatà, N.; Yao, Y.-X.; Wang, C.-Z.; Ho, K.-M.; Kotliar, G. Interplay of spin-orbit and entropic effects in cerium. Phys. Rev. B 2014, 90, 161104. [Google Scholar] [CrossRef] [Green Version]
- Schlottmann, P. Effects of spin-orbit splitting on the ground state of a Ceion. J. Phys. C Solid State Phys. 1985, 18, 1865. [Google Scholar] [CrossRef]
- Jarlborg, T. The behavior of f-levels in hcp and bcc rare-earth elements in the ground state and XPS and BIS spectroscopy from density-functional theory. J. Phys. Condens. Matter 2014, 26, 155503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, B.; Pezzoli, M.E.; Sordi, G.; Haule, K.; Kotliar, G. α-γ transition in cerium: Magnetic form factor and dynamic magnetic susceptibility in dynamical mean-field theory. Phys. Rev. B 2014, 89, 125113. [Google Scholar] [CrossRef]
- Li, R.-S.; Zhou, X.-H.; Zheng, X.-H.; Huang, S.-Q.; Tian, S.-P. Temperature-independent localization of Ce 4f electrons in cerium monoarsenide. Chin. J. Phys. 2022, 75, 215–225. [Google Scholar] [CrossRef]
- Bi, X.; Hu, X.; Li, Q. Effect of Co addition into Ni film on shear strength of solder/Ni/Cu system: Experimental and theoretical investigations. Mater. Sci. Eng. A 2020, 788, 139589. [Google Scholar] [CrossRef]
- Mattsson, A.E.; Wills, J.M. Density functional theory for d- and f-electron materials and compounds. Int. J. Quantum Chem. 2016, 116, 834–846. [Google Scholar] [CrossRef] [Green Version]
- Casadei, M.; Ren, X.; Rinke, P.; Rubio, A.; Scheffler, M. Density-Functional Theory for f-Electron Systems: The α−γ Phase Transition in Cerium. Phys. Rev. Lett. 2012, 109, 146402. [Google Scholar] [CrossRef] [Green Version]
- Sarker, H.P.; Huda, M.N. Role of f-electrons in determining insulator to metal phase transitions of Ca (La1−xCex) 2S4 (0 ≤ x ≤ 1) solid solution: A DFT + U study. J. Appl. Phys. 2021, 130, 145102. [Google Scholar] [CrossRef]
- Yamagami, H.; Mavromaras, A.; Kübler, J. Magnetic properties of f-electron systems in spin-polarized relativistic density functional theory. J. Phys. Condens. Matter 1997, 9, 10881. [Google Scholar] [CrossRef]
- Tolba, S.A.; Gameel, K.M.; Ali, B.A.; Almossalami, H.A.; Allam, N.K. Density Functional Calculations: The DFT + U: Approaches, Accuracy, and Applications; IntechOpen: London, UK, 2018. [Google Scholar]
- van Santen, R.A.; Sautet, P. Computational Methods in Catalysis and Materials Science: An Introduction for Scientists and Engineers; Wiley: New York, NY, USA, 2015. [Google Scholar]
- Haule, K.; Pascut, G.L. Mott Transition and Magnetism in Rare Earth Nickelates and its Fingerprint on the X-ray Scattering. Sci. Rep. 2017, 7, 10375. [Google Scholar] [CrossRef] [Green Version]
- Park, K.; Pascut, G.L.; Khanal, G.; Yokosuk, M.O.; Xu, X.; Gao, B.; Gutmann, M.J.; Litvinchuk, A.P.; Kiryukhin, V.; Cheong, S.-W.; et al. Band-Mott mixing hybridizes the gap in Fe2Mo3O8. Phys. Rev. B 2021, 104, 195143. [Google Scholar] [CrossRef]
- Sterkhov, E.V.; Chtchelkatchev, N.M.; Mostovshchikova, E.V.; Ryltsev, R.E.; Uporov, S.A.; Pascut, G.L.; Fetisov, A.V.; Titova, S.G. The origin of the structural transition in double-perovskite manganite PrBaMn2O6. J. Alloy. Compd. 2022, 892, 162034. [Google Scholar] [CrossRef]
- Stanislavchuk, T.N.; Pascut, G.L.; Litvinchuk, A.P.; Liu, Z.; Sungkyun, C.; Gutmann, M.J.; Gao, B.; Haule, K.; Kiryukhin, V.; Cheong, S.-W.; et al. Spectroscopic and first principle DFT + eDMFT study of complex structural, electronic, and vibrational properties of M2Mo3O8 (M = Fe, Mn) polar magnets. Phys. Rev. B 2020, 102, 115139. [Google Scholar] [CrossRef]
- Koçer, C.P.; Haule, K.; Pascut, G.L.; Monserrat, B. Efficient lattice dynamics calculations for correlated materials with DFT + DMFT. Phys. Rev. B 2020, 102, 245104. [Google Scholar] [CrossRef]
- Pascut, G.L.; Haule, K. Role of Orbital Selectivity on Crystal Structures and Electronic States in Bimno3 and Lamno3 Perovskites. arXiv 2020, arXiv:2005.12179. [Google Scholar]
- Shim, J.H.; Haule, K.; Kotliar, G. Modeling the Localized-to-Itinerant Electronic Transition in the Heavy Fermion System CeIrIn5. Science 2007, 318, 1615–1617. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.H.; Haule, K.; Kotliar, G. Fluctuating valence in a correlated solid and the anomalous properties of δ-plutonium. Nature 2007, 446, 513–516. [Google Scholar] [CrossRef] [Green Version]
- Bosak, A.; Hoesch, M.; Krisch, M.; Chernyshov, D.; Pattison, P.; Schulze-Briese, C.; Winkler, B.; Milman, V.; Refson, K.; Antonangeli, D.; et al. 3D Imaging of the Fermi Surface by Thermal Diffuse Scattering. Phys. Rev. Lett. 2009, 103, 076403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Q.; Wu, L.; Cao, H.; Kang, C.-J.; Nelson, C.; Pascut, G.L.; Besara, T.; Siegrist, T.; Haule, K.; Kotliar, G.; et al. Vacancy defect control of colossal thermopower in FeSb2. NPJ Quantum Mater 2021, 6, 13. [Google Scholar] [CrossRef]
- Hackett, T.A.; Baldwin, D.J.; Paudyal, D. Electronic, magnetic, and magnetocrystalline anisotropy properties of light lanthanides. J. Magn. Magn. Mater. 2017, 441, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Rasaili, P.; Sharma, N.K.; Bhattarai, A. Comparison of Ferromagnetic Materials: Past Work, Recent Trends, and Applications. Condens. Matter 2022, 7, 12. [Google Scholar] [CrossRef]
- Yin, L.; Parker, D.S. Effect of atom substitutions on the magnetic properties in Ce2Fe17: Toward permanent magnet applications. J. Appl. Phys. 2021, 129, 103902. [Google Scholar] [CrossRef]
- Li, L.; Yan, M. Recent progresses in exploring the rare earth based intermetallic compounds for cryogenic magnetic refrigeration. J. Alloy. Compd. 2020, 823, 153810. [Google Scholar] [CrossRef]
- Soleimani, Z.; Zoras, S.; Ceranic, B.; Shahzad, S.; Cui, Y. A review on recent developments of thermoelectric materials for room-temperature applications. Sustain. Energy Technol. Assess. 2020, 37, 100604. [Google Scholar] [CrossRef]
- Finn, P.A.; Asker, C.; Wan, K.; Bilotti, E.; Fenwick, O.; Nielsen, C.B. Thermoelectric Materials: Current Status and Future Challenges. Front. Electron. Mater. 2021, 1, 677845. [Google Scholar] [CrossRef]
- Bessais, L. Structure and Magnetic Properties of Intermetallic Rare-Earth-Transition-Metal Compounds: A Review. Materials 2022, 15, 201. [Google Scholar] [CrossRef] [PubMed]
- Coldea, M.; Pop, V.; Neumann, M.; Isnard, O.; Pascut, L.G. Magnetic properties of Al–Gd–Ni orthorhombic compounds. J. Alloy. Compd. 2005, 390, 16–20. [Google Scholar] [CrossRef]
- Zhang, Y. Review of the structural, magnetic and magnetocaloric properties in ternary rare earth RE2T2X type intermetallic compounds. J. Alloy. Compd. 2019, 787, 1173–1186. [Google Scholar] [CrossRef]
- Coldea, M.; Neumann, M.; Chiuzbaian, S.G.; Pop, V.; Pascut, L.G.; Isnard, O.; Takács, A.F.; Pacurariu, R. X-ray photoelectron spectroscopy and magnetism of Mn–Pd alloys. J. Alloys Compd. 2006, 417, 7–12. [Google Scholar] [CrossRef]
- Buschow, K.H.J. Handbook of Magnetic Materials; Elsevier: Amsterdam, The Netherlands, 2001; Volume 13. [Google Scholar]
- Pascut, G.L.; Birol, T.; Gutmann, M.J.; Yang, J.J.; Cheong, S.-W.; Haule, K.; Kiryukhin, V. Series of alternating states with unpolarized and spin-polarized bands in dimerized IrTe2. Phys. Rev. B 2014, 90, 195122. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model 1 Flat Pd/Sb | Model 2 Split Pd/Sb | Model 3 Puckered Pd/Sb | |
|---|---|---|---|
| LaPdSb | |||
| Space group | P63/mmc | P63/mmc | P63mc |
| a = b [Å] | 4.6134 (10) | 4.6134 (10) | 4.6134 (10) |
| c [Å] | 8.1470 (20) | 8.1470 (20) | 8.1470 (20) |
| V [Å3] | 150.17 (6) | 150.17 (6) | 150.17 (6) |
| La (x, y, z) | (0, 0, 0) | (0, 0, 0) | (0, 0, 0) * |
| U11 = U22[Å2] | 0.0088 (1) | 0.00893 (4) | 0.00898 (2) |
| U33 [Å2] | 0.0072 (1) | 0.00767 (4) | 0.00746 (3) |
| Pd (x, y, z) | (1/3, 2/3, 3/4) | (1/3, 2/3, 0.7380 (5)) | (1/3, 2/3, 0.7429 (4)) |
| U11 = U22[Å2] | 0.0073 (1) | 0.00725 (3) | 0.00822 (3) |
| U33 [Å2] | 0.0240 (3) | 0.0170 (1) | 0.01155 (6) |
| Sb (x, y, z) | (2/3, 1/3, 3/4) | (2/3, 1/3, 0.7591 (5)) | (2/3, 1/3, 0.76424 (6)) |
| U11 = U22[Å2] | 0.0073 (1) ** | 0.00725 (3) ** | 0.00602 (2) |
| U33 [Å2] | 0.0240 (3) ** | 0.0170 (1) ** | 0.02594 (2) |
| NRefs (I > 3σ/all) Neutron X-ray *** | 9273/2687 1278/314 | 9273/2687 1278/314 | 9273/2687 2359/646 |
| R1/wR2 (I > 3σ) | 23.07/64.69 | 8.17/22.05 | 6.49/15.54 |
| R1/wR2 (all) | 24.67/64.73 | 9.39/22.45 | 7.81/16.06 |
| GooF (I > 3σ/all) | 19.97/17.64 | 5.90/5.30 | 4.23/3.85 |
| Model 1 Flat Pd/Sb | Model 2 Split Pd/Sb | Model 3 Puckered Pd/Sb | |
|---|---|---|---|
| CePdSb | |||
| Space group | P63/mmc | P63/mmc | P63mc |
| a = b [Å] | 4.6071 (7) | 4.6071 (7) | 4.6071 (7) |
| c [Å] | 7.9371 (16) | 7.9371 (16) | 7.9371 (16) |
| V [Å3] | 145.90 (4) | 145.90 (4) | 145.90 (4) |
| Ce (x, y, z) | (0, 0, 0) | (0, 0, 0) | (0, 0, 0) * |
| U11 = U22[Å2] | 0.0098 (4) | 0.0103 (1) | 0.00974 (8) |
| U33 [Å2] | 0.0053 (4) | 0.0057 (1) | 0.00687 (8) |
| Pd (x, y, z) | (1/3, 2/3, 3/4) | (1/3, 2/3, 0.7400 (2)) | (1/3, 2/3, 0.7166 (1)) |
| U11 = U22[Å2] | 0.0069 (3) | 0.00719 (7) | 0.0069 (1) |
| U33 [Å2] | 0.060 (1) | 0.01262 (5) | 0.0354 (3) |
| Sb (x, y, z) | (2/3, 1/3, 3/4) | (2/3, 1/3, 0.7824 (3)) | (2/3, 1/3, 0.76295 (7)) |
| U11 = U22[Å2] | 0.0069 (3) ** | 0.00719 (7) ** | 0.0072 (1) |
| U33 [Å2] | 0.060 (1) ** | 0.01262 (5) ** | 0.0117 (1) |
| NRefs (I > 3σ/all) | 6216/6 | 6126/6 | 6216/6 |
| R1/wR2 (I > 3σ) | 41.90/82.02 | 11.61/28.90 | 8.03/19.11 |
| R1/wR2 (all) | 41.91/82.02 | 11.62/28.95 | 8.04/19.18 |
| GooF (I > 3σ/all) | 31.47/31.45 | 8.50/8.50 | 5.58/5.59 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutmann, M.J.; Pascut, G.L.; Katoh, K.; Zimmermann, M.v.; Refson, K.; Adroja, D.T. New Insights on the Electronic-Structural Interplay in LaPdSb and CePdSb Intermetallic Compounds. Materials 2022, 15, 7678. https://doi.org/10.3390/ma15217678
Gutmann MJ, Pascut GL, Katoh K, Zimmermann Mv, Refson K, Adroja DT. New Insights on the Electronic-Structural Interplay in LaPdSb and CePdSb Intermetallic Compounds. Materials. 2022; 15(21):7678. https://doi.org/10.3390/ma15217678
Chicago/Turabian StyleGutmann, Matthias Josef, Gheorghe Lucian Pascut, Kenichi Katoh, Martin von Zimmermann, Keith Refson, and Devashibhai Thakarshibhai Adroja. 2022. "New Insights on the Electronic-Structural Interplay in LaPdSb and CePdSb Intermetallic Compounds" Materials 15, no. 21: 7678. https://doi.org/10.3390/ma15217678