
The Effect of Yttrium on the Solution and Diffusion Behaviors of Helium in Tungsten: First-Principles Simulations

Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
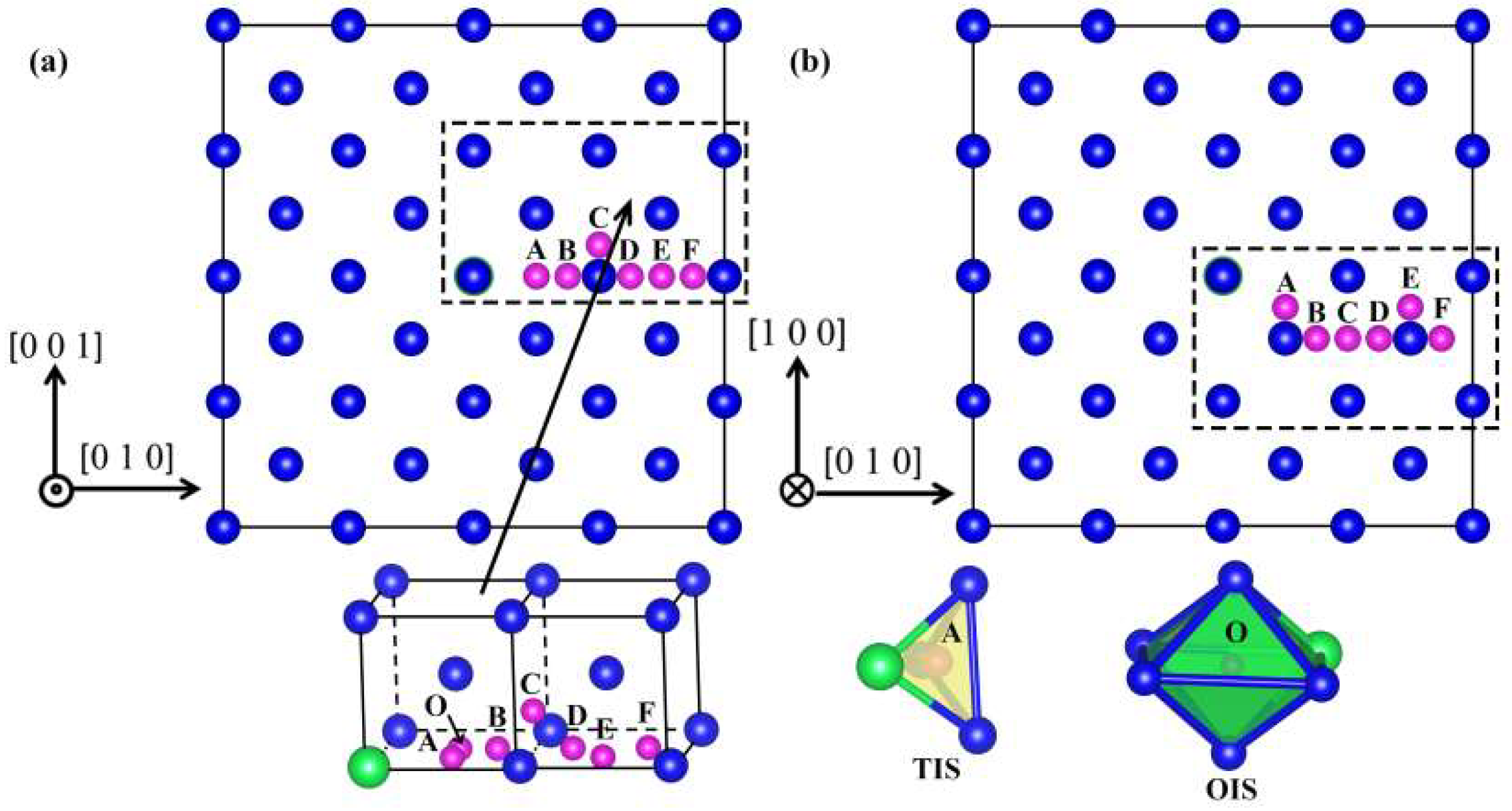
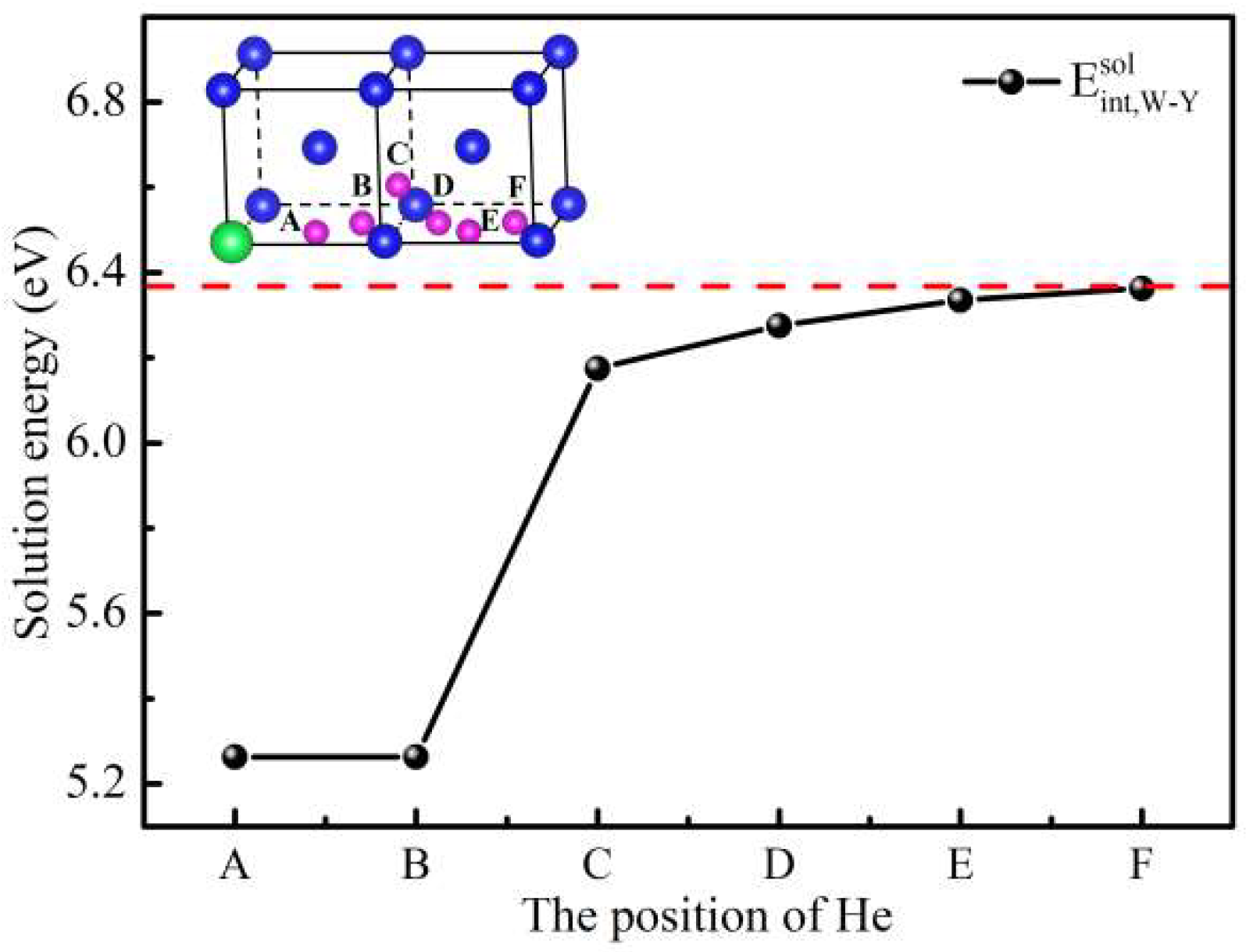
3.1. Effect of Y on Solution Energy of He in W
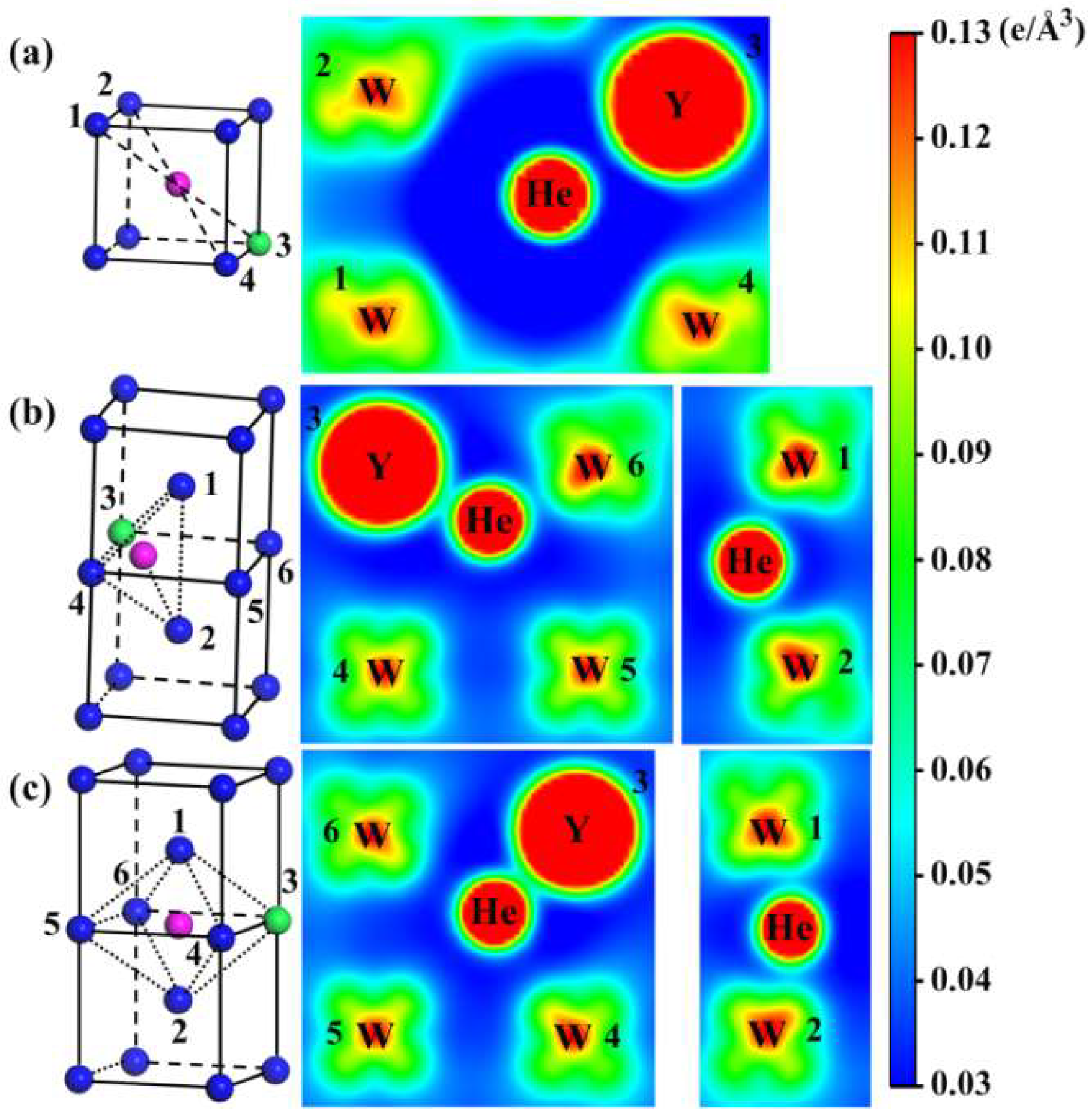
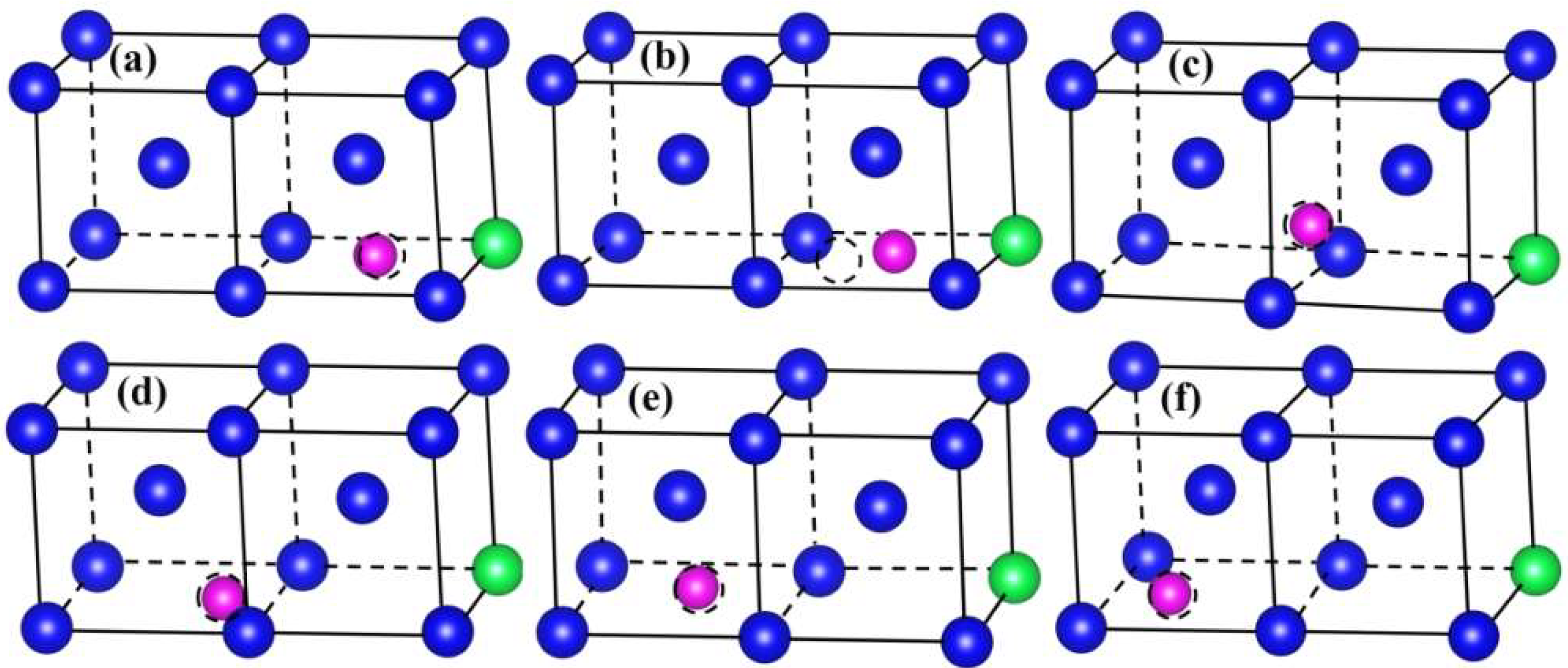
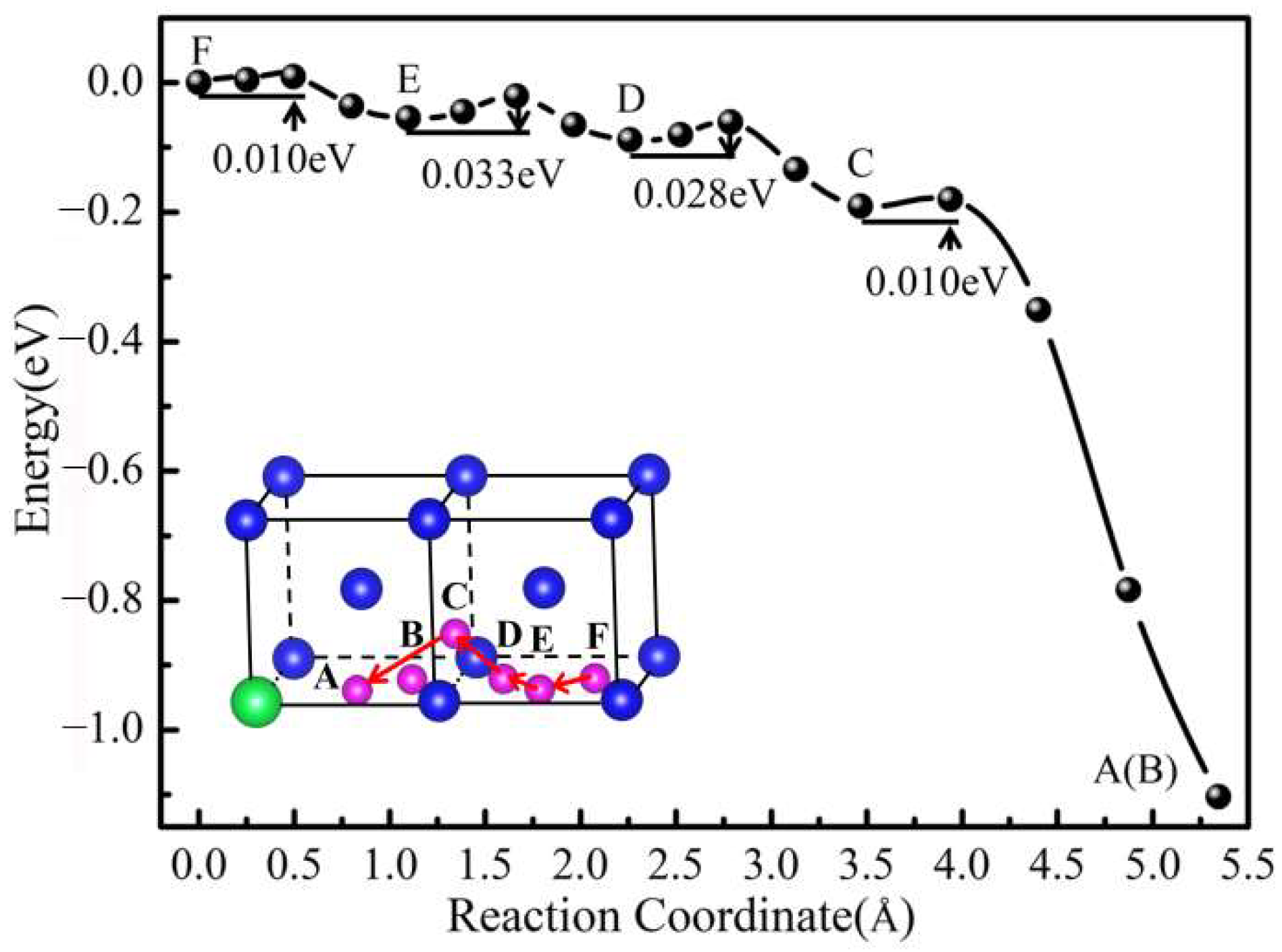
3.2. Interaction between Y and He Atoms
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meade, D. 50 years of fusion research. Nucl. Fusion 2010, 50, 14004. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Li, Y.H.; Zhou, H.B.; Deng, H.Q.; Lu, G.H. Segregation and aggregation of rhenium in tungsten grain boundary: Energetics, configurations and strengthening effects. J. Nucl. Mater. 2020, 528, 151867. [Google Scholar] [CrossRef]
- Barabash, V.; Federici, G.; Rödig, M.; Snead, L.L.; Wu, C.H. Neutron irradiation effects on plasma facing materials. J. Nucl. Mater. 2000, 283–287, 138–146. [Google Scholar] [CrossRef]
- Lee, H.T.; Haasz, A.A.; Davis, J.W.; Macaulay-Newcombe, R.G. Hydrogen and helium trapping in tungsten under single and sequential irradiations. J. Nucl. Mater. 2007, 360, 196–207. [Google Scholar] [CrossRef]
- Jiang, D.Y.; Ouyang, C.Y.; Liu, S.Q. Mechanical properties of W-Ti alloys from first-principles calculations. Fusion Eng. Des. 2016, 106, 34–39. [Google Scholar] [CrossRef]
- Zhou, H.B.; Li, Y.H.; Lu, G.H. Modeling and simulation of helium behavior in tungsten: A first-principles investigation. Comput. Mater. Sci. 2016, 112, 487–491. [Google Scholar] [CrossRef]
- Zhang, N.N.; Zhang, Y.J.; Yang, Y.; Zhang, P.; Ge, C.C. First-principles study of structural, mechanical, and electronic properties of W alloying with Zr. Chin. Phys. B 2019, 28, 046301. [Google Scholar] [CrossRef]
- Zhou, Z.J.; Pintsuk, G.; Linke, J.; Hirai, T.; Rödig, M. Transient high heat load tests on pure ultra-fine grained tungsten fabricated by resistance sintering under ultra-high pressure. Fusion Eng. Des. 2010, 85, 115–121. [Google Scholar] [CrossRef]
- Wirtz, M.; Linke, J.; Pintsuk, G.; Singheiser, L.; Uytdenhouwen, I. Comparison of the thermal shock performance of different tungsten grades and the influence of microstructure on the damage behaviour. Phys. Scr. 2011, T145, 014058. [Google Scholar] [CrossRef]
- Linke, J.; Loewenhoff, T.; Massaut, V.; Pintsuk, G.; Ritz, G.; Rödig, M.; Schmidt, A.; Thomser, C.; Uytdenhouwen, I.; Vasechko, V.; et al. Performance of different tungsten grades under transient thermal loads. Nucl. Fusion 2011, 51, 073017. [Google Scholar] [CrossRef]
- Wu, M.Y.; Zhang, Y.J.; Wang, Z.H.; Qiu, K.K.; Shi, Y.X.; Ge, C.C. Theoretical investigation on yttrium clustering in tungsten grain boundary region and strengthening effect. J. Appl. Phys. 2022, 132, 125106. [Google Scholar] [CrossRef]
- Zhao, M.Y.; Zhou, Z.J.; Ding, Q.M.; Zhong, M.; Tan, J. The investigation of Y doping content effect on the microstructure and microhardness of tungsten materials. Mater. Sci. Eng. A 2014, 618, 572–577. [Google Scholar] [CrossRef]
- Veleva, L.; Oksiuta, Z.; Vogt, U.; Baluc, N. Sintering and characterization of W-Y and W-Y2O3 materials. Fusion Eng. Des. 2009, 84, 1920–1924. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, Y.C.; Yu, F.W.; Chen, J.L. Study on structure and property of tungsten alloy strengthened with dispersed La2O3. Rare Met. Mater. Eng. 2007, 36, 822–824. [Google Scholar]
- Kim, Y.; Lee, K.H.; Kim, E.P.; Cheong, D.I.; Hong, S.H. Fabrication of high temperature oxides dispersion strengthened tungsten composites by spark plasma sintering process. Int. J. Refract. Met. Hard Mater. 2009, 27, 842–846. [Google Scholar] [CrossRef]
- Luo, L.M.; Shi, J.; Zan, X.; Li, P.; Luo, G.N.; Chen, J.L.; Wu, Y.C. Current status and development trend on alloying elements-doped plasma-facing tungsten-based materials. Chin. J. Nonferrous Met. 2016, 26, 1899–1911. [Google Scholar]
- Li, X.C.; Lu, T.; Pan, X.D.; Xu, Y.P.; Niu, G.J.; Xu, Q.; Zhou, H.S.; Luo, G.N. Molecular dynamics study of the hydrogen and helium interaction in tungsten. Nucl. Instrum. Methods Phys. Res. Sect. B 2019, 455, 114–117. [Google Scholar] [CrossRef]
- Wang, S.W.; Guo, W.G.; Yuan, Y.; Gao, N.; Zhu, X.L.; Cheng, L.; Cao, X.Z.; Fu, E.G.; Shi, L.Q.; Gao, F.; et al. Evolution of vacancy defects in heavy ion irradiated tungsten exposed to helium plasma. J. Nucl. Mater. 2020, 532, 152051. [Google Scholar] [CrossRef]
- Yue, F.Y.; Li, Y.H.; Ren, Q.Y.; Ma, F.F.; Li, Z.Z.; Zhou, H.B.; Deng, H.Q.; Zhang, Y.; Lu, G.H. Suppressing/enhancing effect of rhenium on helium clusters evolution in tungsten: Dependence on rhenium distribution. J. Nucl. Mater. 2021, 543, 152545. [Google Scholar] [CrossRef]
- Trinkaus, H.; Singh, B.N. Helium accumulation in metals during irradiation–where do we stand. J. Nucl. Mater. 2003, 323, 229–242. [Google Scholar] [CrossRef]
- Ullmaier, H. The influence of helium on the bulk properties of fusion reactor structural materials. Nucl. Fusion 1984, 24, 1039–1083. [Google Scholar] [CrossRef]
- Philipps, V. Tungsten as material for plasma-facing components in fusion devices. J. Nucl. Mater. 2011, 415, S2–S9. [Google Scholar] [CrossRef]
- Ueda, Y.; Schmid, K.; Balden, M.; Coenen, J.W.; Loewenhoff, T.; Ito, A.; Hasegawa, A.; Hardie, C.; Porton, M.; Gilbert, M. Baseline high heat flux and plasma facing materials for fusion. Nucl. Fusion 2017, 57, 92006. [Google Scholar] [CrossRef] [Green Version]
- Brooks, J.N.; Allain, J.P.; Doerner, R.P.; Hassanein, A.; Nygren, R.; Rognlien, T.D.; Whyte, D.G. Plasma-surface interaction issues of an all-metal ITER. Nucl. Fusion 2009, 49, 35007. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.N.; Zhang, Y.J.; Yang, Y.; Zhang, P.; Ge, C.C. Effects of Zr and V additions on the stability and migration of He in bcc W: A first-principles study. Phys. Lett. A 2019, 383, 2777–2783. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.N.; Zhang, Y.J.; Yang, Y.; Zhang, P.; Hu, Z.Y.; Ge, C.C. Trapping of helium atom by vacancy in tungsten: A density functional theory study. Eur. Phys. J. B 2017, 90, 101. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17959. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillonin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Zhang, N.N.; Zhang, Y.J.; Zhang, P.; Yang, Y.; Hu, Z.Y.; Ge, C.C. Theoretical insight into the effects of nitrogen and vacancy defects on the behavior of helium in tungsten. Appl. Phys. Express 2018, 11, 15801. [Google Scholar] [CrossRef]
- Wu, M.Y.; Wang, Z.H.; Zhang, N.N.; Ge, C.C.; Zhang, Y.J. Theoretical predictions of the structural and mechanical properties of tungsten–rare earth element alloys. Materials 2021, 14, 3046. [Google Scholar] [CrossRef] [PubMed]
- Grimes, R.W.; Catlow, C.R.A. The stability of fission products in uranium dioxide. Philos. Trans. R. Soc. A-Math. Phys. Eng. Sci. 1991, 335, 609–634. [Google Scholar] [CrossRef]
- Seletskaia, T.; Osetsky, Y.; Stoller, R.E.; Stocks, G.M. First-principles theory of the energetics of He defects in bcc transition metals. Phys. Rev. B 2008, 78, 134103. [Google Scholar] [CrossRef]
- Song, C.; Hou, J.; Kong, X.S.; Chen, L.; Wang, S.; Liu, C.S. Structures and energetics of multiple helium atoms in a tungsten monovacancy. J. Nucl. Mater. 2022, 561, 153577. [Google Scholar] [CrossRef]
- Wu, M.Y.; Lv, W.T.; Zhang, Y.J.; Yang, Y.; Wang, Z.H.; Qiu, K.K.; Shi, Y.X.; Zhao, B.; Ge, C.C. Potassium clusters in tungsten grain boundaries: Formation mechanism and strengthening effect. J. Nucl. Mater. 2022, 573, 154135. [Google Scholar] [CrossRef]
- Yan, W.L.; Zhou, H.B.; Jin, S.; Zhang, Y.; Lu, G.H. Dissolution energetics and its strain dependence of transition metal alloying elements in tungsten. J. Nucl. Mater. 2015, 456, 260–265. [Google Scholar] [CrossRef]
- Becquart, C.S.; Domain, C. Migration energy of He in W revisited by ab initio calculations. Phys. Rev. Lett. 2006, 97, 196402. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; You, Y.; Liu, L.; Fan, H.; Ni, W.; Liu, D.; Liu, C.S.; Benstetter, G.; Wang, Y. Nanostructured fuzz growth on tungsten under low-energy and high-flux He irradiation. Sci. Rep. 2015, 5, 10959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.B.; Ou, X.; Zhang, Y.; Shu, X.L.; Liu, Y.L.; Lu, G.H. Effect of carbon on helium trapping in tungsten: A first-principles investigation. J. Nucl. Mater. 2013, 440, 338–343. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Sub | TIS | OIS | ||
|---|---|---|---|---|---|
| Bulk W | Solution energy (eV) | 4.809 | 6.367 | 6.510 | Present |
| 4.770 a | 6.32 b | 6.56 b | Reference | ||
| 0.039 | 0.047 | 0.050 | Error intervals | ||
| Bader charge (e) | 2.139 | 2.159 | 2.165 | Present | |
| Bulk W-Y | Solution energy (eV) | 4.670 | 5.263 | 5.362 | Present |
| Bader charge (e) | 2.128 | 2.156 | 2.160 | Present |
| Site | A | B | C | D | E | F |
|---|---|---|---|---|---|---|
| Solution energy (eV) | 5.263 | 5.263 | 6.175 | 6.275 | 6.335 | 6.363 |
| Binding energy (eV) | 1.029 | 1.029 | 0.117 | 0.017 | 0.008 | 0.003 |
| Bader charge (e) | 2.156 | 2.156 | 2.160 | 2.161 | 2.171 | 2.173 |
| Distance (Å) | 1.926 | 1.926 | 3.507 | 4.241 | 4.860 | 5.729 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, M.; Zhang, Y.; Qiu, K.; Shi, Y.; Jin, J.; Ge, C. The Effect of Yttrium on the Solution and Diffusion Behaviors of Helium in Tungsten: First-Principles Simulations. Materials 2022, 15, 8468. https://doi.org/10.3390/ma15238468
Wu M, Zhang Y, Qiu K, Shi Y, Jin J, Ge C. The Effect of Yttrium on the Solution and Diffusion Behaviors of Helium in Tungsten: First-Principles Simulations. Materials. 2022; 15(23):8468. https://doi.org/10.3390/ma15238468
Chicago/Turabian StyleWu, Mingyu, Yujuan Zhang, Kaikai Qiu, Yaxian Shi, Jingyuan Jin, and Changchun Ge. 2022. "The Effect of Yttrium on the Solution and Diffusion Behaviors of Helium in Tungsten: First-Principles Simulations" Materials 15, no. 23: 8468. https://doi.org/10.3390/ma15238468