
Comparison of Physical/Chemical Properties of Prussian Blue Thin Films Prepared by Different Pulse and DC Electrodeposition Methods
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
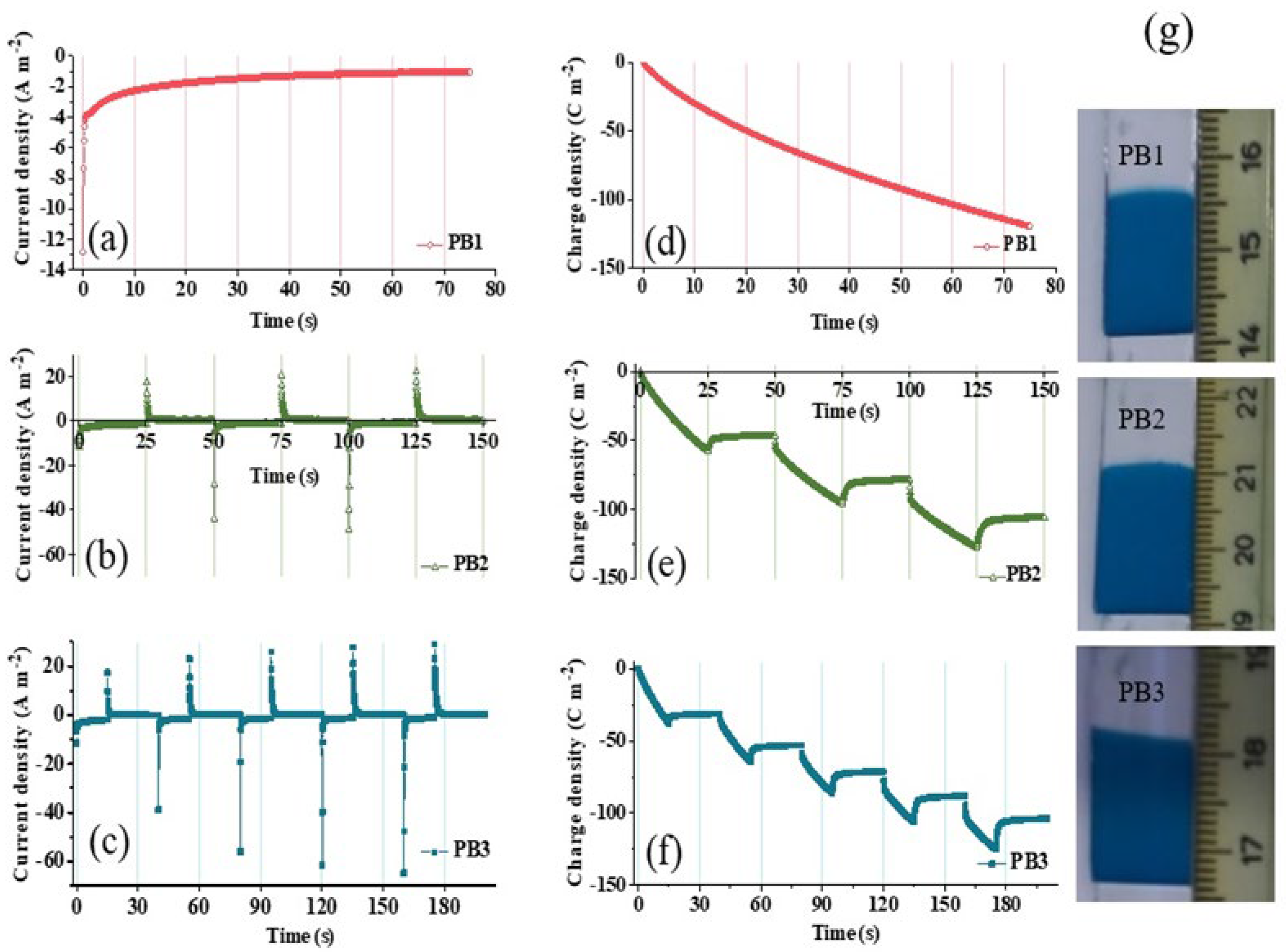
3.1. Electrodeposition Process
3.2. Morphological Analysis
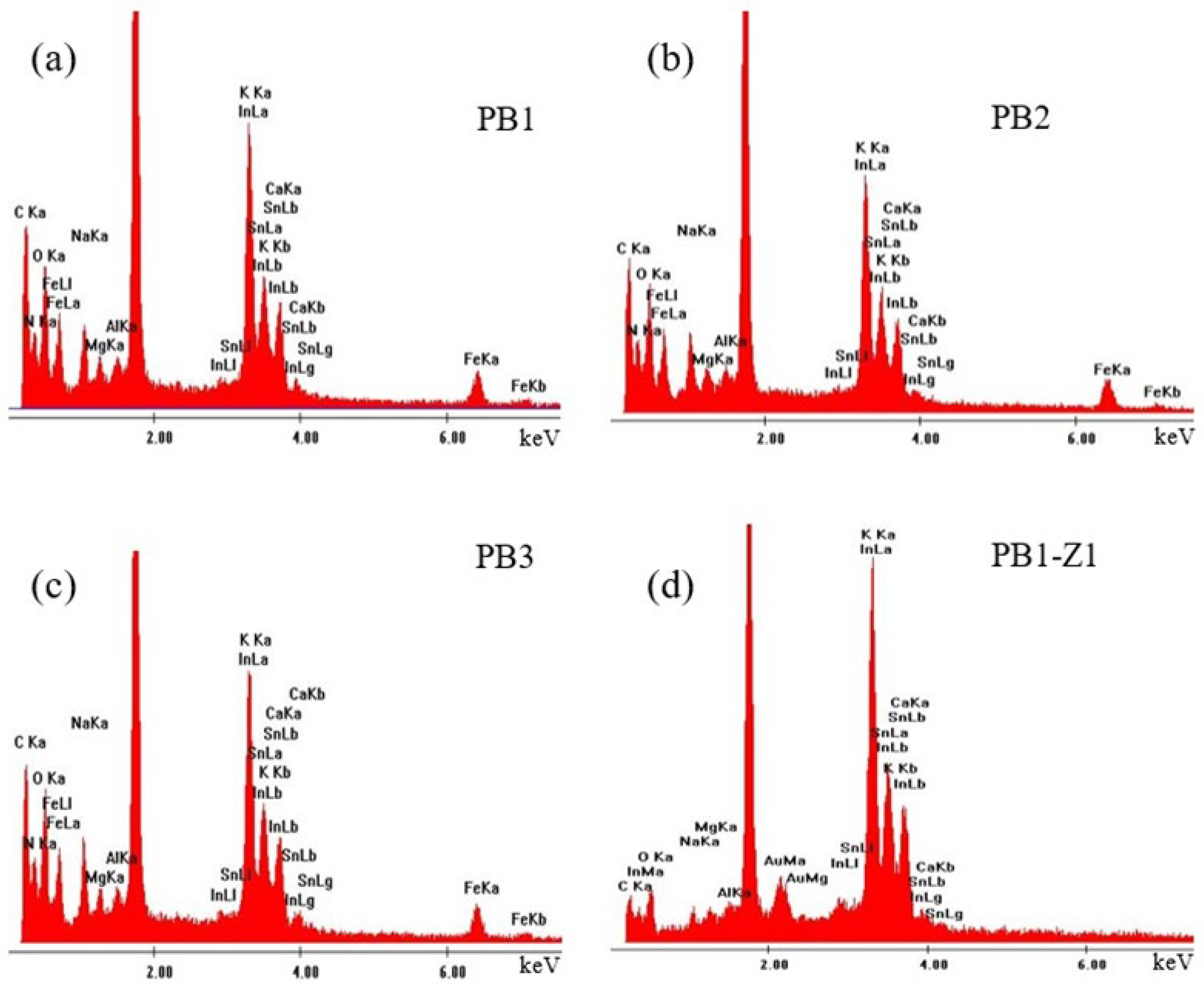
3.3. Elemental Analysis of the Samples
3.4. FTIR-ATR Analysis
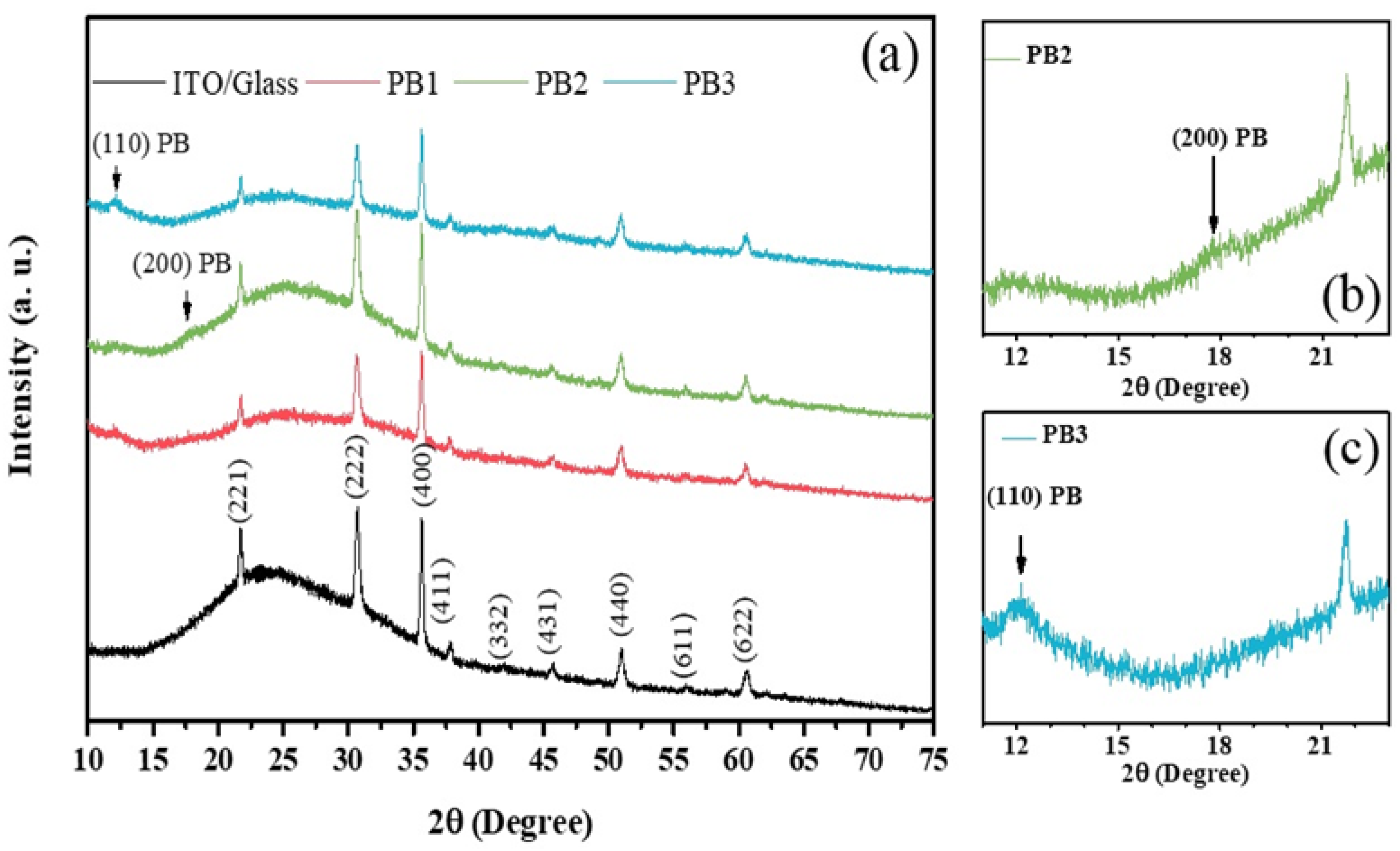
3.5. Structural Analysisof the PB Films
3.6. UV-Vis Spectroscopic Analysis
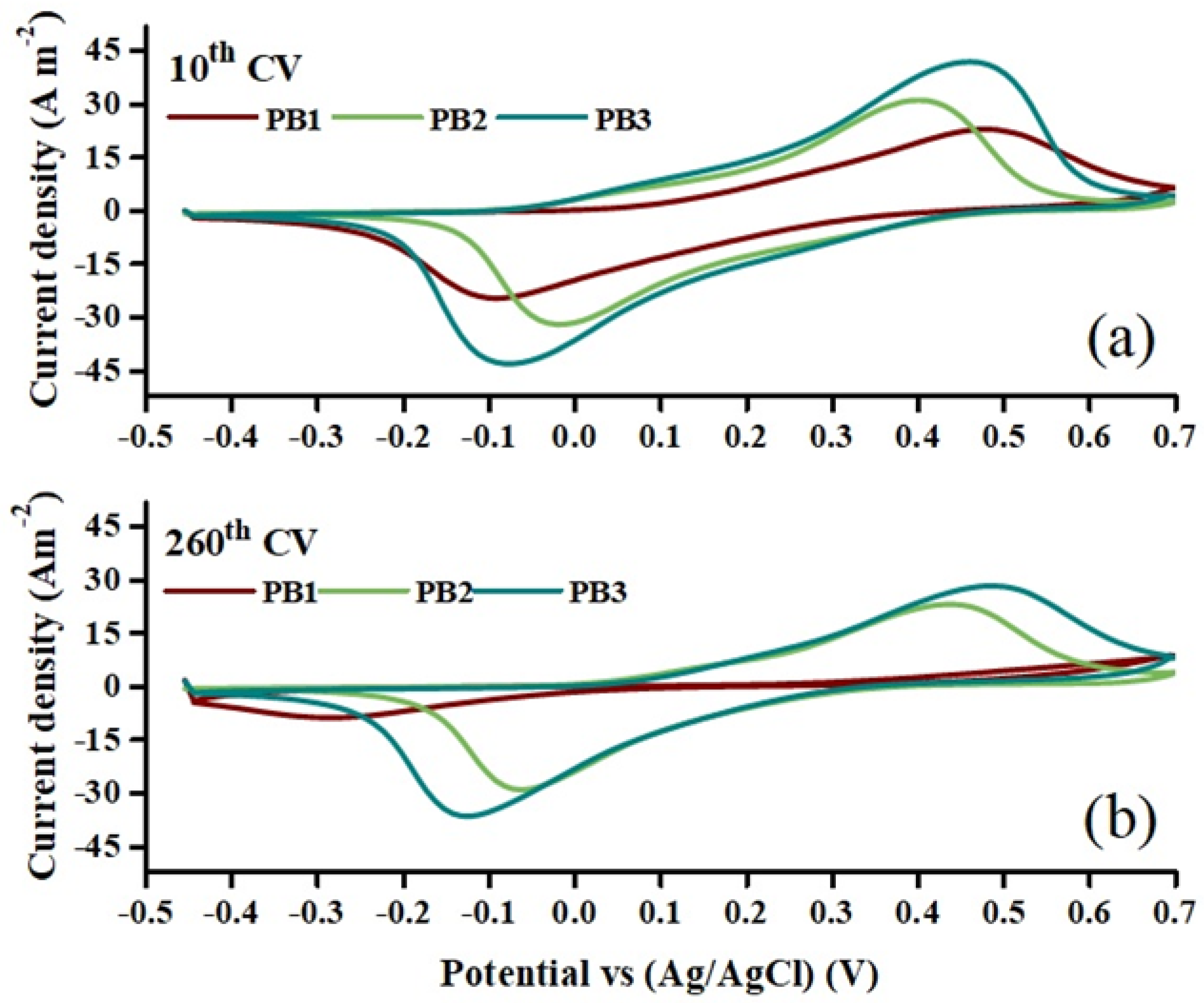
3.7. Cyclic Voltammetry Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Monk, P.M.; Mortimer, R.J.; Rosseinsky, D.R. Electrochromism and Electrochromic Devices; Cambridge University Press: Cambridge, UK, 2007; Volume 421. [Google Scholar]
- Isfahani, V.B.; Memarian, N.; Dizaji, H.R.; Arab, A.; Silva, M.M. The physical and electrochromic properties of Prussian Blue thin films electrodeposited on ITO electrodes. Electrochim. Acta 2019, 304, 282–291. [Google Scholar] [CrossRef]
- Isfahani, V.B.; Pereira, R.F.; Fernandes, M.; Sabadini, R.C.; Pereira, S.; Dizaji, H.R.; Arab, A.; Fortunato, E.; Pawlicka, A.; Rego, R. Gellan-Gum and LiTFSI-Based Solid Polymer Electrolytes for Electrochromic Devices. ChemistrySelect 2021, 6, 5110–5119. [Google Scholar] [CrossRef]
- Patnaik, S.G.; Pech, D. Low Temperature Deposition of Highly Cyclable Porous Prussian Blue Cathode for Lithium-Ion Microbattery. Small 2021, 17, 2101615. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Qin, R.; Ding, S.; Wang, Y.; Li, S.; Zhao, Q.; Pan, F. Structure and properties of prussian blue analogues in energy storage and conversion applications. Adv. Funct. Mater. 2021, 31, 2006970. [Google Scholar] [CrossRef]
- Chu, Z.; Liu, Y.; Jin, W. Recent progress in Prussian blue films: Methods used to control regular nanostructures for electrochemical biosensing applications. Biosens. Bioelectron. 2017, 96, 17–25. [Google Scholar] [CrossRef]
- de Tacconi, N.R.; Rajeshwar, K.; Lezna, R.O. Metal hexacyanoferrates: Electrosynthesis, in situ characterization, and applications. Chem. Mater. 2003, 15, 3046–3062. [Google Scholar] [CrossRef]
- He, Y.; Zhang, P.; Wang, F.; Wang, L.; Su, Y.; Zhang, F.; Zhuang, X.; Feng, X. Vacancy modification of Prussian-blue nano-thin films for high energy-density micro-supercapacitors with ultralow RC time constant. Nano Energy 2019, 60, 8–16. [Google Scholar] [CrossRef]
- Qiu, M.; Sun, P.; Liu, Y.; Huang, Q.; Zhao, C.; Li, Z.; Mai, W. Visualized UV photodetectors based on prussian blue/TiO2 for smart irradiation monitoring application. Adv. Mater. Technol. 2018, 3, 1700288. [Google Scholar] [CrossRef]
- Lin, X.; Cao, Y.; Xue, Y.; Wu, F.; Yu, F.; Wu, M.; Zhu, X. Multifunctional theranostic agents based on prussian blue nanoparticles for tumor targeted and MRI—guided photodynamic/photothermal combined treatment. Nanotechnology 2020, 31, 135101. [Google Scholar] [CrossRef]
- Yang, R.; Hou, M.; Gao, Y.; Zhang, L.; Xu, Z.; Kang, Y.; Xue, P. Indocyanine green-modified hollow mesoporous Prussian blue nanoparticles loading doxorubicin for fluorescence-guided tri-modal combination therapy of cancer. Nanoscale 2019, 11, 5717–5731. [Google Scholar] [CrossRef]
- Rosseinsky, D.R.; Glidle, A. EDX, spectroscopy, and composition studies of electrochromic iron (III) hexacyanoferrate (II) deposition. J. Electrochem. Soc. 2003, 150, C641. [Google Scholar] [CrossRef]
- Rosseinsky, D.R.; Glasser, L.; Jenkins, H.D.B. Thermodynamic clarification of the curious ferric/potassium ion exchange accompanying the electrochromic redox reactions of Prussian blue, iron (III) hexacyanoferrate (II). J. Am. Chem. Soc. 2004, 126, 10472–10477. [Google Scholar] [CrossRef] [PubMed]
- García-Jareño, J.; Navarro-Laboulais, J.; Vicente, F. A numerical approach to the voltammograms of the reduction of Prussian Blue films on ITO electrodes. Electrochim. Acta 1997, 42, 1473–1480. [Google Scholar] [CrossRef]
- Giménez-Romero, D.; Agrisuelas, J.; García-Jareño, J.J.; Gregori, J.; Gabrielli, C.; Perrot, H.; Vicente, F. Electromechanical phase transition in hexacyanometallate nanostructure (Prussian blue). J. Am. Chem. Soc. 2007, 129, 7121–7126. [Google Scholar] [CrossRef]
- Lundgren, C.; Murray, R.W. Observations on the composition of Prussian blue films and their electrochemistry. Inorg. Chem. 1988, 27, 933–939. [Google Scholar] [CrossRef]
- Mamontova, E.; Daurat, M.; Long, J.; Godefroy, A.; Salles, F.; Guari, Y.; Gary-Bobo, M.; Larionova, J. Fashioning Prussian blue nanoparticles by adsorption of luminophores: Synthesis, properties, and in vitro imaging. Inorg. Chem. 2020, 59, 4567–4575. [Google Scholar] [CrossRef]
- Cahu, M.; Ali, L.M.; Sene, S.; Long, J.; Camerel, F.; Ciancone, M.; Salles, F.; Chopineau, J.; Devoisselle, J.-M.; Felix, G. A rational study of the influence of Mn 2+-insertion in Prussian blue nanoparticles on their photothermal properties. J. Mater. Chem. B 2021, 9, 9670–9683. [Google Scholar] [CrossRef]
- Yu, Z.-Y.; Duan, Y.; Liu, J.-D.; Chen, Y.; Liu, X.-K.; Liu, W.; Ma, T.; Li, Y.; Zheng, X.-S.; Yao, T. Unconventional CN vacancies suppress iron-leaching in Prussian blue analogue pre-catalyst for boosted oxygen evolution catalysis. Nat. Commun. 2019, 10, 2799. [Google Scholar] [CrossRef] [Green Version]
- Grandjean, F.; Samain, L.; Long, G.J. Characterization and utilization of Prussian blue and its pigments. Dalton Trans. 2016, 45, 18018–18044. [Google Scholar] [CrossRef]
- Bueno, P.R.; Ferreira, F.F.; Giménez-Romero, D.; Oliveira Setti, G.; Faria, R.C.; Gabrielli, C.; Perrot, H.; Garcia-Jareño, J.J.; Vicente, F. Synchrotron structural characterization of electrochemically synthesized hexacyanoferrates containing K+: A revisited analysis of electrochemical redox. J. Phys. Chem. C 2008, 112, 13264–13271. [Google Scholar] [CrossRef]
- Mamontova, E. Multifunctional Nanomaterials BASED on Coordination Networks. Ph.D. Thesis, Université Montpellier, Montpellier, France, 2020. [Google Scholar]
- Brown, D.B. Mixed-Valence Compounds: Theory and Applications in Chemistry, Physics, Geology, and Biology; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; Volume 58. [Google Scholar]
- Agrisuelas, J.; García-Jareño, J.J.; Gimenez-Romero, D.; Vicente, F. Insights on the mechanism of insoluble-to-soluble Prussian blue transformation. J. Electrochem. Soc. 2009, 156, P149. [Google Scholar] [CrossRef]
- Boergert, M.J. Crystal Structure, Thermal Expansion, and Water Occupancies in Prussian Blue Analogs: A Combined Neutron, X-ray, and Microscopy Study; New Mexico State University: Las Cruces, NM, USA, 2016. [Google Scholar]
- Zhang, W.; Song, H.; Cheng, Y.; Liu, C.; Wang, C.; Khan, M.A.N.; Zhang, H.; Liu, J.; Yu, C.; Wang, L. Core–shell prussian blue analogs with compositional heterogeneity and open cages for oxygen evolution reaction. Adv. Sci. 2019, 6, 1801901. [Google Scholar] [CrossRef] [PubMed]
- Demiri, S.; Najdoski, M.; Velevska, J. A simple chemical method for deposition of electrochromic Prussian blue thin films. Mater. Res. Bull. 2011, 46, 2484–2488. [Google Scholar] [CrossRef]
- Koncki, R.; Lenarczuk, T.; Radomska, A.; Głąb, S. Optical biosensors based on Prussian Blue films. Analyst 2001, 126, 1080–1085. [Google Scholar] [CrossRef]
- Agrisuelas, J.; García-Jareño, J.J.; Vicente, F. Identification of processes associated with different iron sites in the Prussian Blue structure by in situ electrochemical, gravimetric, and spectroscopic techniques in the dc and ac regimes. J. Phys. Chem. C 2012, 116, 1935–1947. [Google Scholar] [CrossRef]
- Karyakin, A.A. Prussian blue and its analogues: Electrochemistry and analytical applications. Electroanal. Int. J. Devoted Fundam. Pract. Asp. Electroanal. 2001, 13, 813–819. [Google Scholar] [CrossRef]
- Duek, E.A.; De Paoli, M.A.; Mastragostino, M. A solid-state electrochromic device based on polyaniline, prussian blue and an elastomeric electrolyte. Adv. Mater. 1993, 5, 650–652. [Google Scholar] [CrossRef]
- Elshorbagy, M.; Ramadan, R.; Abdelhady, K. Preparation and characterization of spray-deposited efficient Prussian blue electrochromic thin film. Optik 2017, 129, 130–139. [Google Scholar] [CrossRef]
- Isfahani, V.B.; Dizaji, H.R.; Memarian, N.; Arab, A. Electrodeposition of Prussian Blue films: Study of deposition time effect on electrochemical properties. Mater. Res. Express 2019, 6, 096449. [Google Scholar] [CrossRef]
- Jelle, B.P.; Hagen, G. Performance of an electrochromic window based on polyaniline, prussian blue and tungsten oxide. Sol. Energy Mater. Sol. Cells 1999, 58, 277–286. [Google Scholar] [CrossRef]
- Agnihotry, S.; Singh, P.; Joshi, A.G.; Singh, D.; Sood, K.; Shivaprasad, S. Electrodeposited Prussian blue films: Annealing effect. Electrochim. Acta 2006, 51, 4291–4301. [Google Scholar] [CrossRef]
- Abbaspour, A.; Kamyabi, M.A. Electrochemical formation of Prussian blue films with a single ferricyanide solution on gold electrode. J. Electroanal. Chem. 2005, 584, 117–123. [Google Scholar] [CrossRef]
- Cisternas, R.; Muñoz, E.; Henríquez, R.; Córdova, R.; Kahlert, H.; Hasse, U.; Scholz, F. Irreversible electrostatic deposition of Prussian blue from colloidal solutions. J. Solid State Electrochem. 2011, 15, 2461–2468. [Google Scholar] [CrossRef]
- Najafisayar, P.; Bahrololoom, M. Pulse electrodeposition of Prussian Blue thin films. Thin Solid Film. 2013, 542, 45–51. [Google Scholar] [CrossRef]
- Zolotukhina, E.; Bezverkhyy, I.; Vorotyntsev, M. One-stage periodical anodic-cathodic double pulse deposition of nanocomposite materials. Application to Prussian Blue/polypyrrole film coated electrodes. Electrochim. Acta 2014, 122, 247–258. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Fundamentals and applications. Electrochem. Methods 2001, 2, 580–632. [Google Scholar]
- Baggio, B.F.; Vicente, C.; Pelegrini, S.; Pla Cid, C.C.; Brandt, I.S.; Tumelero, M.A.; Pasa, A.A. Morphology and structure of electrodeposited Prussian Blue and Prussian white thin films. Materials 2019, 12, 1103. [Google Scholar] [CrossRef] [Green Version]
- Kulesza, P.J.; Malik, M.A.; Denca, A.; Strojek, J. In situ FT-IR/ATR spectroelectrochemistry of Prussian blue in the solid state. Anal. Chem. 1996, 68, 2442–2446. [Google Scholar] [CrossRef]
- Kulesza, P.J.; Faszynska, M. Indium (III)-hexacyanof errate as a novel polynuclear mixed-valent inorganic material for preparation of thin zeolitic films on conducting substrates. J. Electroanal. Chem. Interfacial Electrochem. 1988, 252, 461–466. [Google Scholar] [CrossRef]
- Jiang, M.; Zhao, Z. A novel stable electrochromic thin film: A Prussian Blue analogue based on palladium hexacyanoferrate. J. Electroanal. Chem. Interfacial Electrochem. 1990, 292, 281–287. [Google Scholar] [CrossRef]
- Christensen, P.A.; Hamnett, A.; Higgins, S.J. A study of electrochemically grown prussian blue films using Fourier-transform infra-red spectroscopy. J. Chem. Soc. Dalton Trans. 1990, 2233–2238. [Google Scholar] [CrossRef]
- Thirumoorthi, M.; Thomas Joseph Prakash, J. Structure, optical and electrical properties of indium tin oxide ultra thin films prepared by jet nebulizer spray pyrolysis technique. J. Asian Ceram. Soc. 2016, 4, 124–132. [Google Scholar] [CrossRef] [Green Version]
- Rey, J.; Plivelic, T.; Rocha, R.; Tadokoro, S.; Torriani, I.; Muccillo, E. Synthesis of In2O3nanoparticles by thermal decomposition of a citrate gel precursor. J. Nanoparticle Res. 2005, 7, 203–208. [Google Scholar] [CrossRef]
- Nyguist, R.; Kagel, R. Infrared Spectra of Inorganic Compounds; Academic Press: New York, NY, USA, 1971; p. 95. [Google Scholar]
- Karpagavinayagam, P.; Vedhi, C. Green synthesis of iron oxide nanoparticles using Avicennia marina flower extract. Vacuum 2019, 160, 286–292. [Google Scholar] [CrossRef]
- Farah, A.M.; Thema, F.T.; Dikio, E.D. Electrochemical detection of hydrogen peroxide based on graphene oxide/prussian blue modified glassy carbon electrode. Int. J. Electrochem. Sci 2012, 7, 5069–5083. [Google Scholar]
- Lin, L.; Huang, X.; Wang, L.; Tang, A. Synthesis, characterization and the electrocatalytic application of prussian blue/titanate nanotubes nanocomposite. Solid State Sci. 2010, 12, 1764–1769. [Google Scholar] [CrossRef]
- Barbosa, J.; Gomes, I.; Pereira, M.; Moura, C.; Mendes, J.; Almeida, B. Structural and dielectric properties of laser ablated BaTiO3 films deposited over electrophoretically dispersed CoFe2O4 grains. J. Appl. Phys. 2014, 116, 164112. [Google Scholar] [CrossRef]
- Araujo, C.; Almeida, B.; Aguiar, M.; Mendes, J. Structural and magnetic properties of CoFe2O4 thin films deposited by laser ablation on Si (001) substrates. Vacuum 2008, 82, 1437–1440. [Google Scholar] [CrossRef]
- Cullity, B. Elements of X-ray Diffraction; Addison-Wesley Publishing Company Inc.: Boston, MA, USA, 1956. [Google Scholar]
- Yu, Z.; Perera, I.R.; Daeneke, T.; Makuta, S.; Tachibana, Y.; Jasieniak, J.J.; Mishra, A.; Bäuerle, P.; Spiccia, L.; Bach, U. Indium tin oxide as a semiconductor material in efficient p-type dye-sensitized solar cells. NPG Asia Mater. 2016, 8, e305. [Google Scholar] [CrossRef]
- Robin, M.B. The color and electronic configurations of Prussian blue. Inorg. Chem. 1962, 1, 337–342. [Google Scholar] [CrossRef]
- Souto, M.F.; Cukiernik, F.D.; Forlano, P.; Olabe, J.A. Electronic structure of cyano-bridged dinuclear iron complexes. J. Coord. Chem. 2001, 54, 343–353. [Google Scholar] [CrossRef]
- Raghavender, A.; Samantilleke, A.; Sa, P.; Almeida, B.; Vasilevskiy, M.; Hong, N.H. Simple way to make Anatase TiO2 films on FTO glass for promising solar cells. Mater. Lett. 2012, 69, 59–62. [Google Scholar] [CrossRef]
- Cardona, M.; Peter, Y.Y. Fundamentals of Semiconductors; Springer: Berlin/Heidelberg, Germany, 2005; Volume 619. [Google Scholar]
- Bayzi Isfahani, V.; Silva, M.M. Fundamentals and Advances of Electrochromic Systems: A Review. Adv. Eng. Mater. 2021, 23, 2100567. [Google Scholar] [CrossRef]
- Mortimer, R.J.; Reynolds, J.R. In situ colorimetric and composite coloration efficiency measurements for electrochromic Prussian blue. J. Mater. Chem. 2005, 15, 2226–2233. [Google Scholar]
- Talagaeva, N.V.; Zolotukhina, E.V.; Pisareva, P.A.; Vorotyntsev, M.A. Electrochromic properties of Prussian blue–polypyrrole composite films in dependence on parameters of synthetic procedure. J. Solid State Electrochem. 2016, 20, 1235–1240. [Google Scholar] [CrossRef]
- Herren, F.; Fischer, P.; Ludi, A.; Hälg, W. Neutron diffraction study of Prussian Blue, Fe4 [Fe (CN) 6] 3. xH2O. Location of water molecules and long-range magnetic order. Inorg. Chem. 1980, 19, 956–959. [Google Scholar] [CrossRef]
- Ogura, K.; Nakayama, M.; Nakaoka, K. Electrochemical quartz crystal microbalance and in situ infrared spectroscopic studies on the redox reaction of Prussian blue. J. Electroanal. Chem. 1999, 474, 101–106. [Google Scholar] [CrossRef]
- Lovrić, M.; Hermes, M.; Scholz, F. Solid state electrochemical reactions in systems with miscibility gaps. J. Solid State Electrochem. 2000, 4, 394–401. [Google Scholar] [CrossRef]
- Scholz, F.; Dostal, A. The formal potentials of solid metal hexacyanometalates. Angew. Chem. Int. Ed. Engl. 1996, 34, 2685–2687. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | PB1 | PB2 | PB3 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Method | DC CHA | Symmetric Pulse | Non-Symmetric Pulse | |||||||||
| Parameters | VR (V) | TR (s) | VR (V) | TR (s) | VO (V) | TO (s) | Cycles Number | VR (V) | TR (s) | VO (V) | TO (s) | Cycles Number |
| Values | 0.445 | 75 | 0.445 | 25 | 0.860 | 25 | 3 | 0.445 | 15 | 0.860 | 25 | 5 |
| EDX | C (at %) | N (at %) | O (at %) | K (at %) | Fe (at %) |
|---|---|---|---|---|---|
| PB1 | 28.22 | 33.12 | 30.40 | 3.78 | 4.48 |
| PB2 | 27.23 | 34.49 | 29.87 | 3.39 | 5.01 |
| PB3 | 26.76 | 34.45 | 30.96 | 3.36 | 4.46 |
| Sample | PB1 | PB2 | PB3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameters | QAnodic (C m−2) | QCathodic (C m−2) | QAnodic (C m−2) | QCathodic (C m−2) | QAnodic (C m−2) | QCathodic (C m−2) | |||
| Cycle Number | |||||||||
| 2 | 81 | 87 | 89 | 100 | 139 | 152 | |||
| 10 | 77 | 81 | 92 | 96 | 134 | 141 | |||
| 260 | 14 | 17 | 71 | 73 | 93 | 99 | |||
| Parameters | QTotal (C m−2) | χ (%) | QTotal (C m−2) | χ (%) | QTotal (C m−2) | χ (%) | |||
| Cycle Number | R2 | R10 | R2 | R0 | R2 | R0 | |||
| 2 | 169 | 82 | 81 | 189 | 24 | 23 | 291 | 39 | 30 |
| 10 | 158 | 187 | 274 | ||||||
| 260 | 31 | 144 | 192 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bayzi Isfahani, V.; Arab, A.; Horta Belo, J.; Pedro Araújo, J.; Manuela Silva, M.; Gonçalves Almeida, B. Comparison of Physical/Chemical Properties of Prussian Blue Thin Films Prepared by Different Pulse and DC Electrodeposition Methods. Materials 2022, 15, 8857. https://doi.org/10.3390/ma15248857
Bayzi Isfahani V, Arab A, Horta Belo J, Pedro Araújo J, Manuela Silva M, Gonçalves Almeida B. Comparison of Physical/Chemical Properties of Prussian Blue Thin Films Prepared by Different Pulse and DC Electrodeposition Methods. Materials. 2022; 15(24):8857. https://doi.org/10.3390/ma15248857
Chicago/Turabian StyleBayzi Isfahani, Vahideh, Ali Arab, João Horta Belo, João Pedro Araújo, Maria Manuela Silva, and Bernardo Gonçalves Almeida. 2022. "Comparison of Physical/Chemical Properties of Prussian Blue Thin Films Prepared by Different Pulse and DC Electrodeposition Methods" Materials 15, no. 24: 8857. https://doi.org/10.3390/ma15248857