
Formation and Dissociation of CH4 and CO2 Hydrates in Presence of a Sediment Composed by Pure Quartz Mixed with Ti23 Particles



Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Apparatus
2.2. Materials
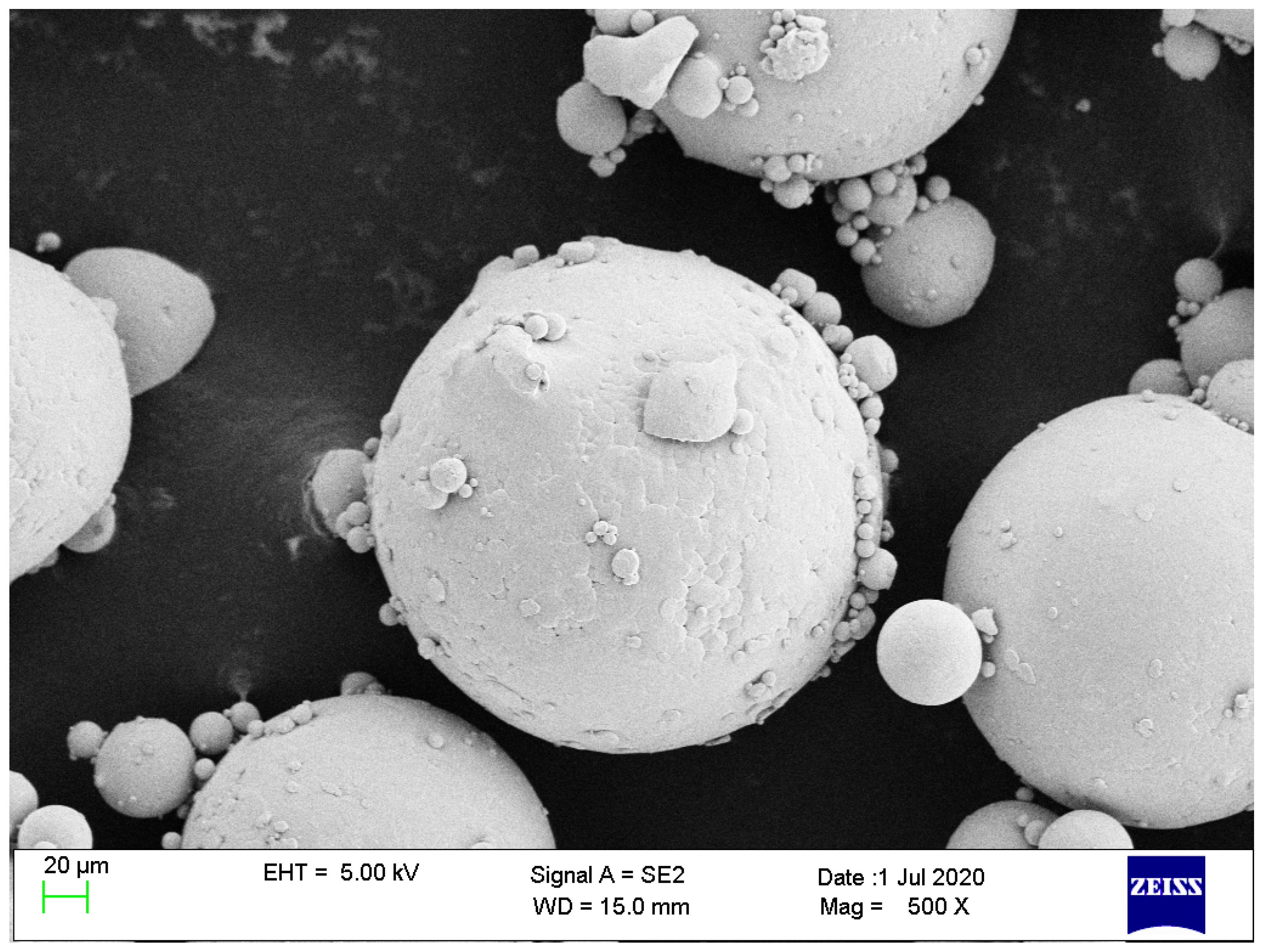
2.2.1. Ti23 Particles
2.3. Methods
3. Results and Discussion
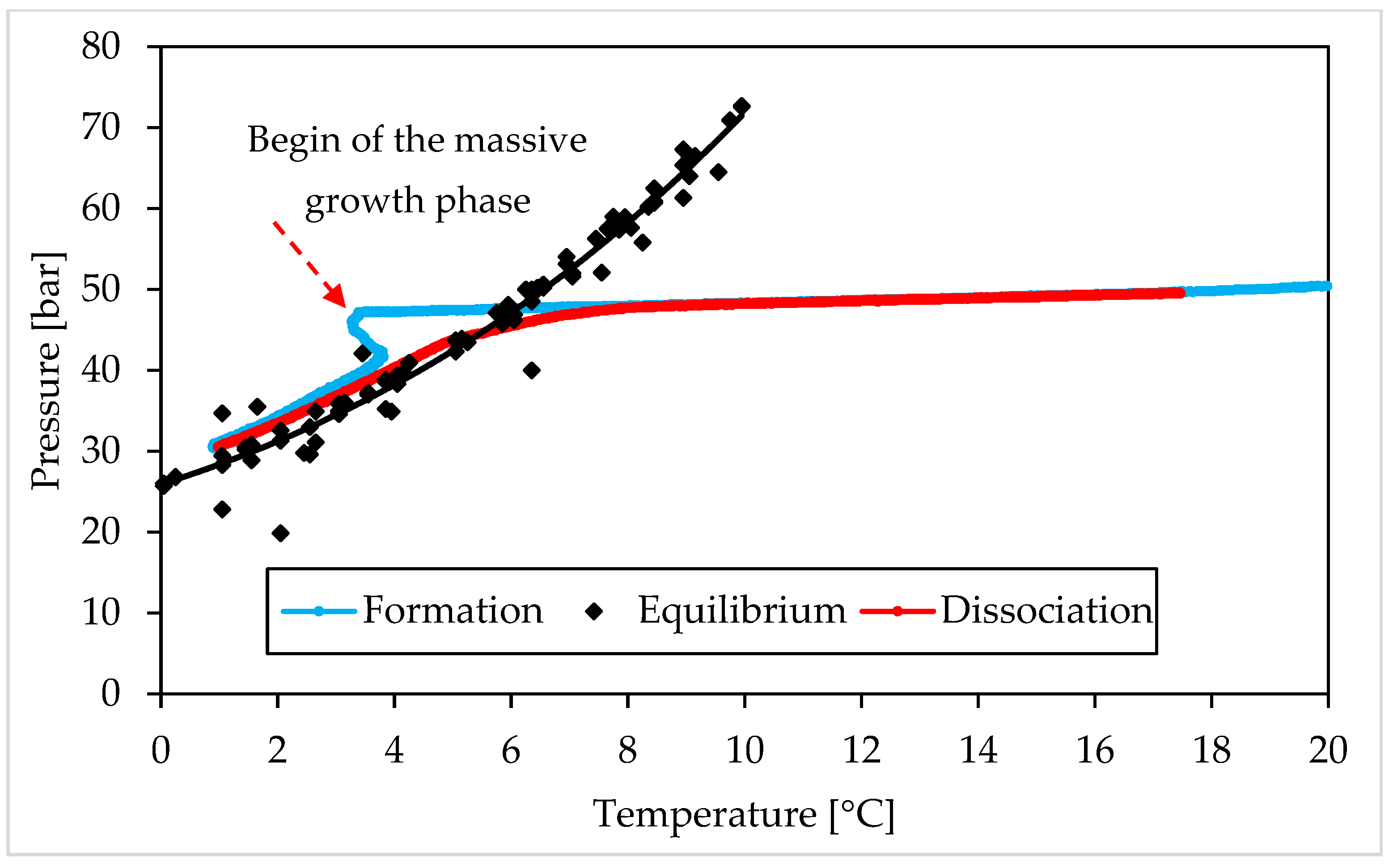
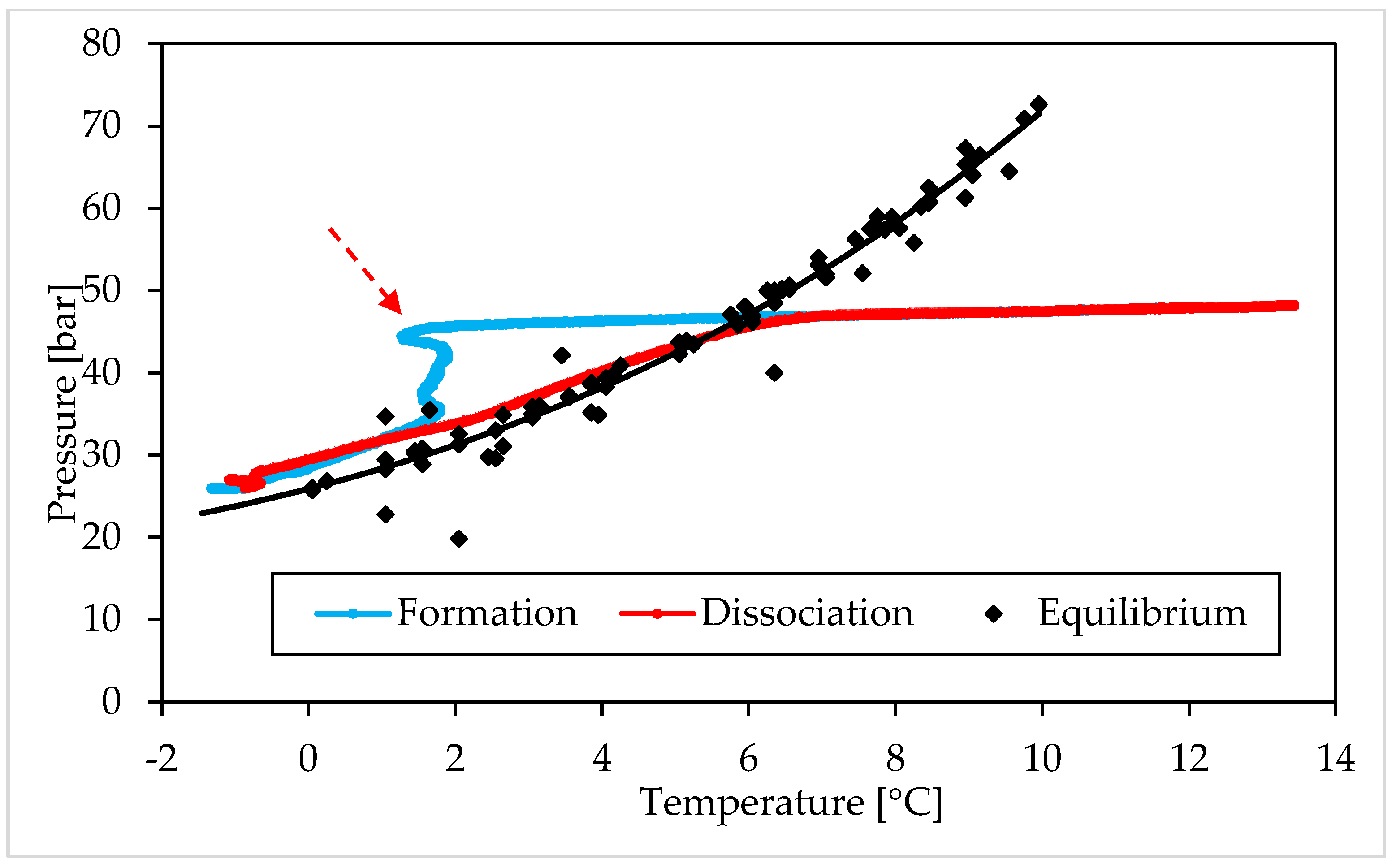
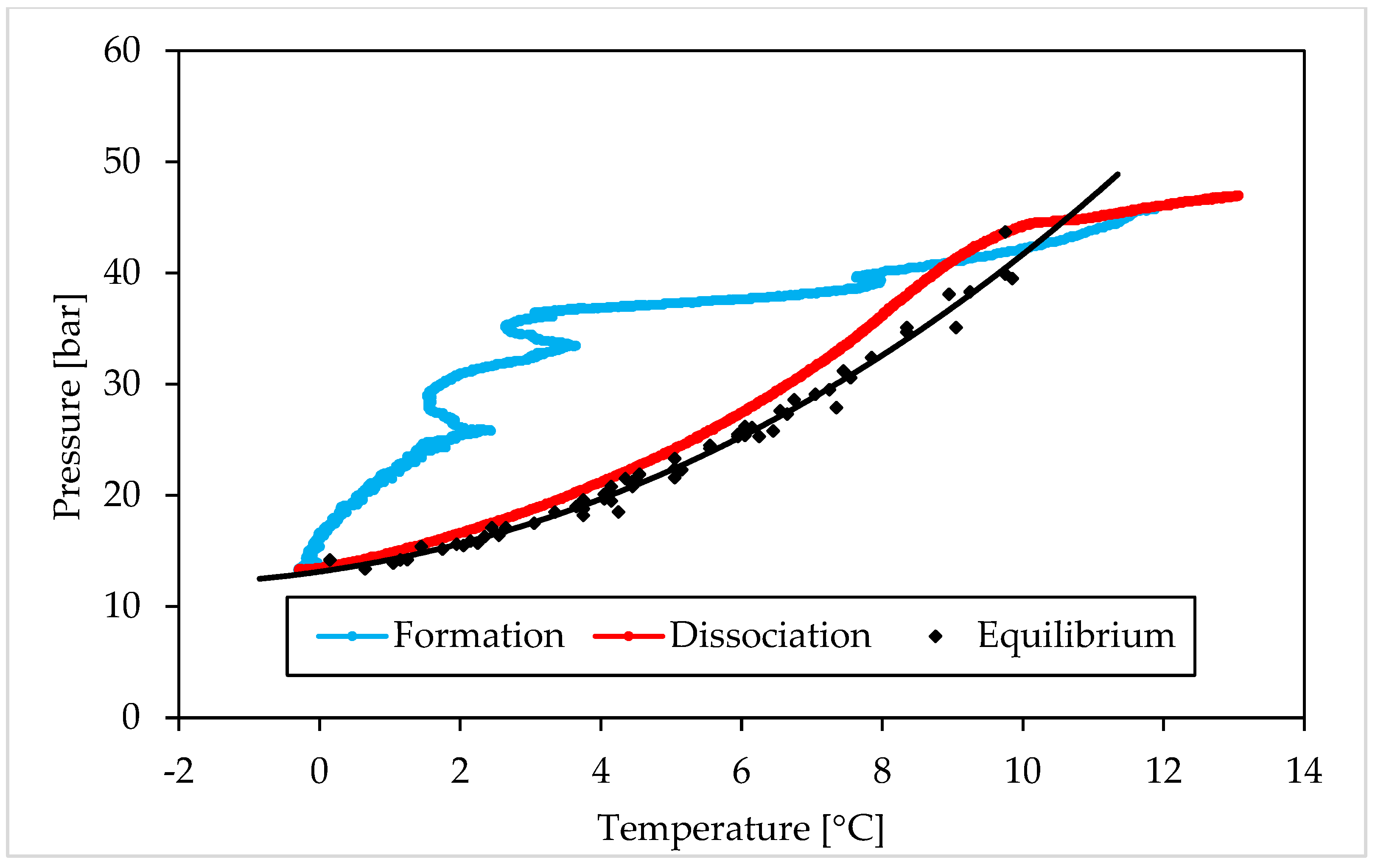
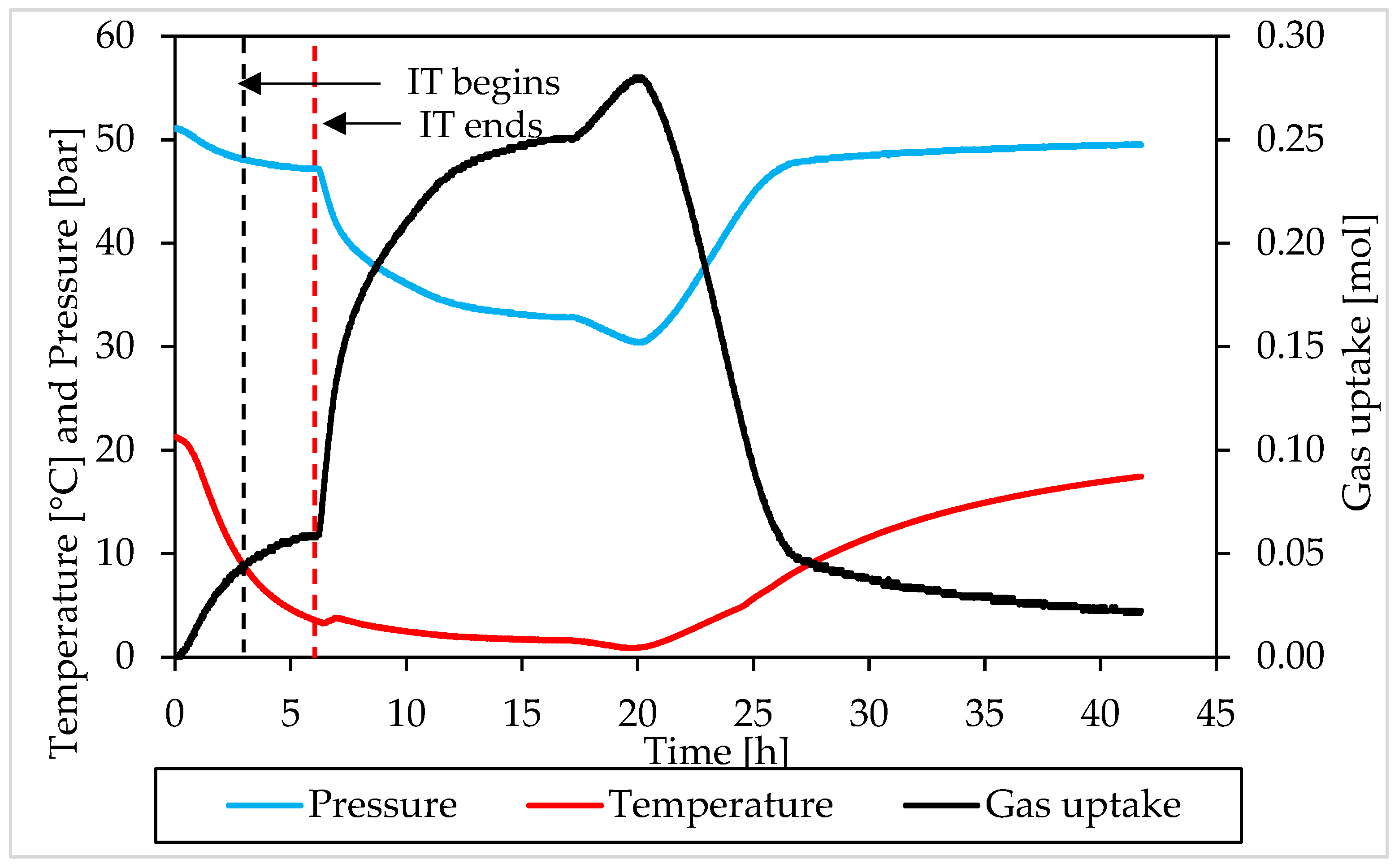
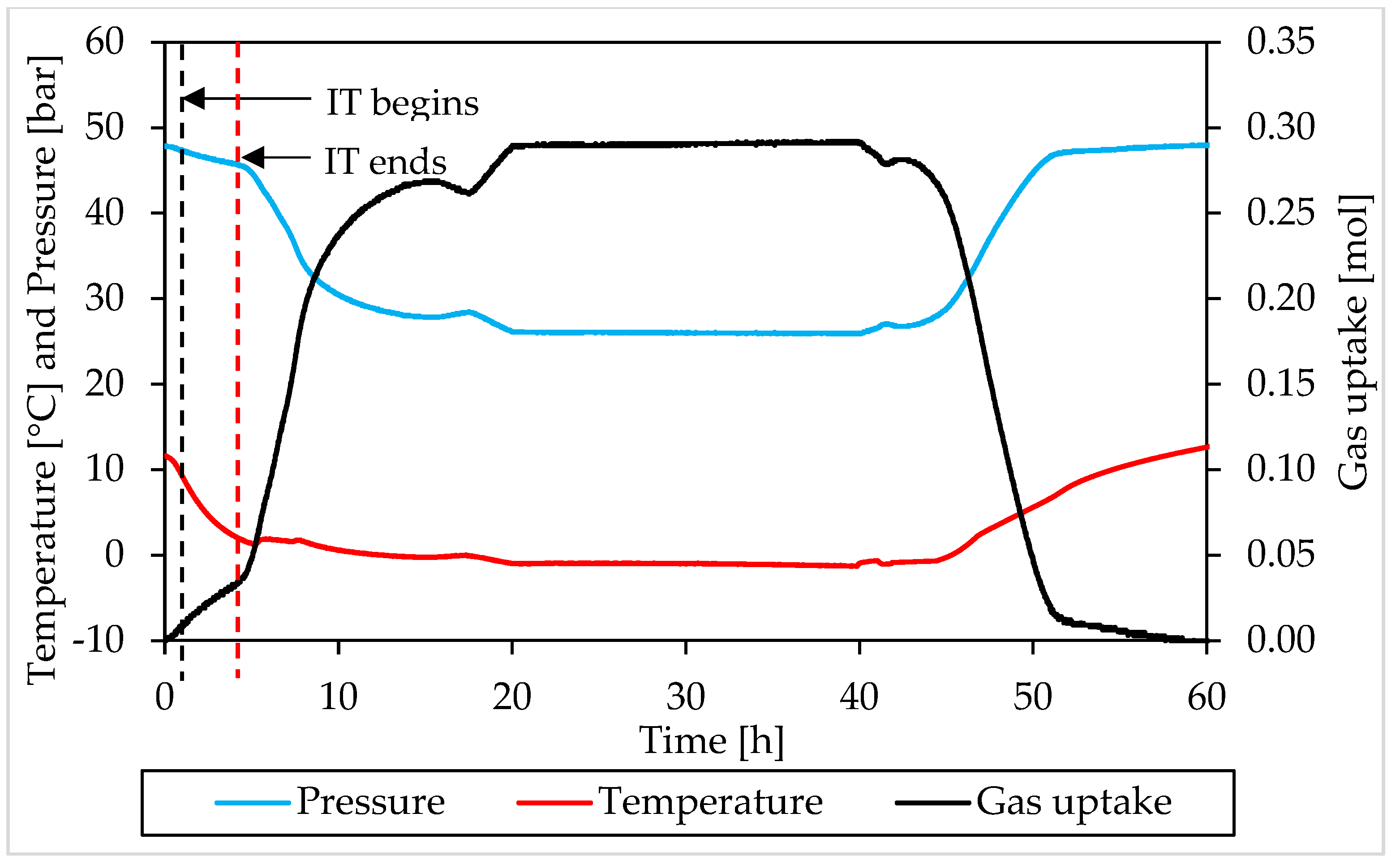
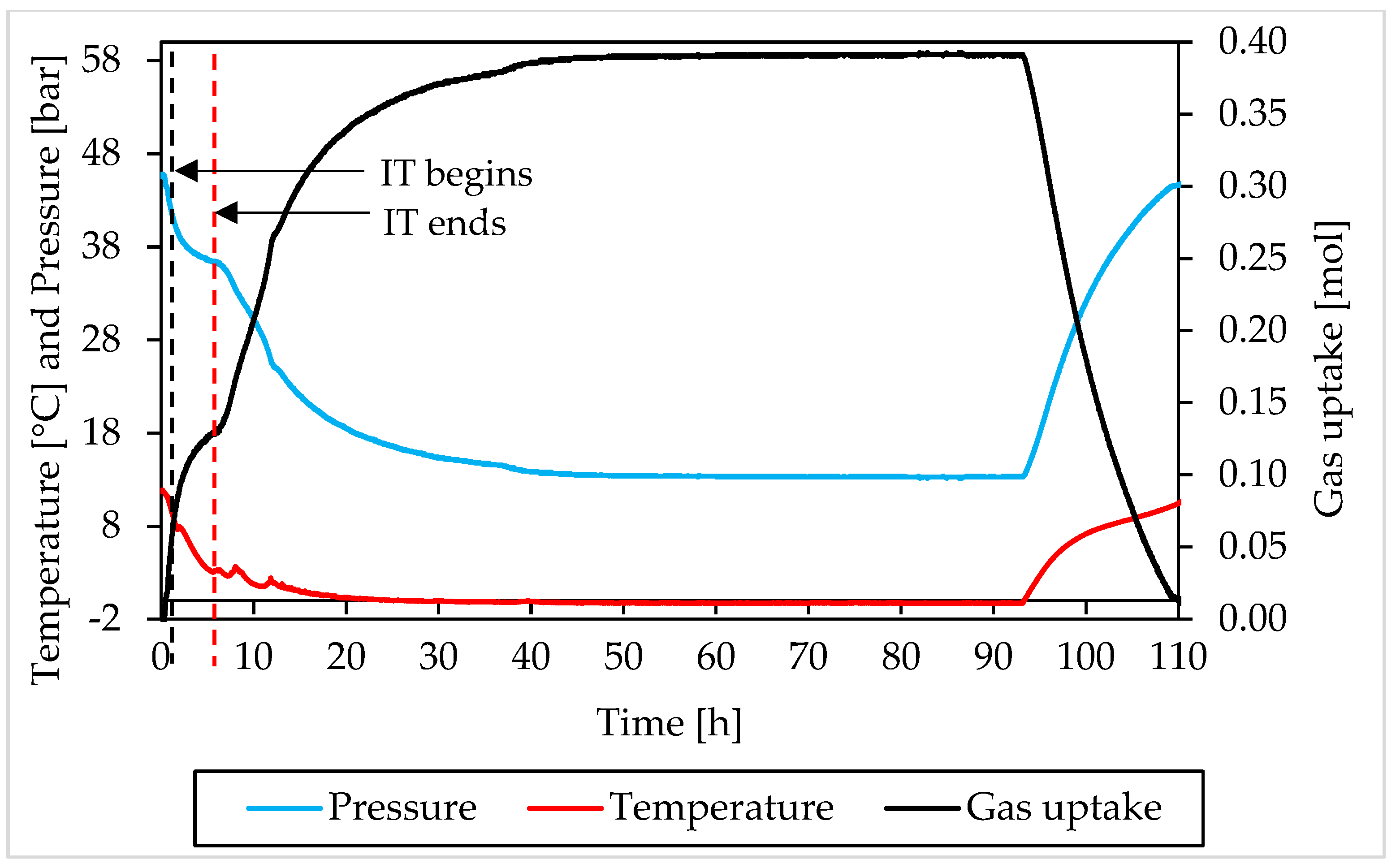
3.1. Pressure and Temperature Evolution Observed during Hydrates Formation and Dissociation
- (i)
- The Ti23 powder, used as additive in this work, did not intervene on the pressure and temperature dissociation values;
- (ii)
- The previous condition did not change with the concentration of such additive.
3.2. Pressure, Temperature and Gas Uptake Evolution over Time
4. Conclusions
- (i)
- The addition of Ti23 particles to the system did not cause relevant changes in the thermodynamic of the process: in all tests and independently from the gaseous guest considered, the dissociation curve approached the phase equilibrium diagram during the whole process;
- (ii)
- In addition, during hydrates formation, none difference in the thermodynamic behavior, as a function of the quantity of Ti23 used, was noticed;
- (iii)
- Similarly, the analyses carried out in this work, did not highlight any relation between the presence of Ti23 (and its concentration) and the quantity of hydrates formed;
- (iv)
- The induction time was found to be different, using the same apparatus, between methane and carbon dioxide hydrates. However, the presence of Ti23 was found to significantly decrease this time period. Moreover, results suggested that this effect is proportional to the concentration of Ti23 in the system. The accelerating effect exercised by Ti23 on the initial portion of hydrates formation appeared clearly and was confirmed in all experiments. In conclusion, the results, produced and described in this work, led us to define that Ti23 powder is a neutral additive for the thermodynamics of methane and carbon dioxide hydrates. Furthermore, it is capable of reducing the time lapse from when the system enters in the hydrate stability zone to when the massive growth phase occurs.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Englezos, P. Clathrate hydrates. Ind. Eng. Chem. Res. 1993, 32, 1251–1274. [Google Scholar] [CrossRef]
- Sloan, E.D. Clathrate Hydrates of Natural Gases; Taylor & Francis: London, UK, 2007. [Google Scholar]
- Rossi, F.; Gambelli, A.M. Thermodynamic phase equilibrium of single—Guest hydrate formation data of hydrate in presence of chemical additives: A review. Fluid Phase Equilib. 2021, 536, 112958. [Google Scholar] [CrossRef]
- Li, X.S.; Xu, C.G.; Zhang, Y.; Ruan, X.K.; Li, G.; Wang, L. Investigation into gas production from natural gas hydrate: A review. Appl. Energy 2016, 172, 286–322. [Google Scholar] [CrossRef] [Green Version]
- Moridis, G.J.; Sloan, E.D. Gas production potential of disperse low-saturation hydrate accumulations in oceanic sediments. Energy Convers. Manag. 2007, 48, 1834–1849. [Google Scholar] [CrossRef] [Green Version]
- Gambelli, A.M.; Presciutti, A.; Rossi, F. Review on the characteristics and advantages related to the use of flue-gas as CO2/N2 mixture for gas hydrate production. Fluid Phase Equilib. 2021, 541, 113077. [Google Scholar] [CrossRef]
- Collett, T.S.; Ginsburg, G.D. Gas hydrates in the Messoyakha gas field of the west Siberian basin—A re-examination of the geologic evidence. Int. J. Offshore Polar Eng. 1997, 8, 96–103. [Google Scholar]
- Collett, T.S.; Kuuskraa, V.A. Hydrates contain vast store of world gas resources. Oil Gas J. 1997, 96, 90–95. [Google Scholar]
- Chen, Z.Y.; Feng, J.C.; Li, X.S.; Zhang, Y.; Li, B.; Lv, Q.N. Preparation of warm brine in situ seafloor based on the hydrate process for marine gas hydrate thermal stimulation. Ind. Eng. Chem. Res. 2014, 53, 14142–14157. [Google Scholar] [CrossRef]
- Li, G.; Moridis, G.J.; Zhang, K.N.; Li, X.S. Evaluation of gas production potential from marine gas hydrate deposits in Shenhu Area of South China Sea. Energy Fuels 2020, 24, 6018–6033. [Google Scholar] [CrossRef]
- Li, X.S.; Wan, L.H.; Li, G.; Li, Q.P.; Chen, Z.Y.; Yan, K.F. Experimental investigation into the production behavior of methane hydrate in porous sediment with hot brine stimulation. Ind. Eng. Chem. Res. 2008, 47, 9696–9702. [Google Scholar] [CrossRef]
- Li, B.; Li, G.; Li, X.S.; Chen, Z.Y.; Zhang, Y. The use of heat-assisted antigravity drainage method in the two horizontal wells in gas production from the Qilian Mountain permafrost hydrate deposits. J. Pet. Sci. Eng. 2014, 120, 141–153. [Google Scholar] [CrossRef]
- Feng, J.C.; Li, X.S.; Li, G.; Li, B.; Chen, Z.Y.; Wang, Y. Numerical investigation of hydrate dissociation performance in the South China Sea with different horizontal well configurations. Energies 2014, 7, 4813–4834. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.C.; Li, G.; Li, X.S.; Li, B.; Chen, Z.Y. Evolution of hydrate dissociation by warm brine stimulation combined depressurization in the South China Sea. Energies 2013, 6, 5402–5425. [Google Scholar] [CrossRef]
- Li, Y.; Gambelli, A.M.; Rossi, F.; Mei, S. Effect of promoters on CO2 hydrate formation: Thermodynamic assessment and microscale Raman spectroscopy/hydrate crystal morphology characterization analysis. Fluid Phase Equilib. 2021, 550, 113218. [Google Scholar] [CrossRef]
- Li, Y.; Chen, J.; Gambelli, A.M.; Zhao, X.; Gao, Y.; Rossi, F.; Mei, S. In situ experimental study on the effect of mixed inhibitors on the phase equilibrium of carbon dioxide hydrate. Chem. Eng. Sci. 2022, 248, 117230. [Google Scholar] [CrossRef]
- Pahlavanzadeh, H.; Nouri, S.; Mohammadi, A.H.; Manteghian, M. Experimental measurements and thermodynamic modelling of hydrate dissociation conditions in CO2 + THF + NaCl + water systems. J. Chem. Thermodyn. 2020, 141, 105956. [Google Scholar] [CrossRef]
- Tsirakis, F.; Stringari, P.; von Solms, N.; Coquelet, C.; Kontogeorgis, G. Hydrate equilibrium data for the CO2 + N2 system with the use of tetra-n-butylammonium bromide (TBAB), cyclopentane (CP) and their mixture. Fluid Phase Equilib. 2016, 408, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Partoon, B.; Wong, N.M.S.; Sabil, K.M.; Nasrifar, K.; Ahmad, M.R. A study on thermodynamics effect of [EMIM]-Cl and [OH-C2MIM]-Cl on methane hydrate equilibrium line. Fluid Phase Equilib. 2013, 337, 26–31. [Google Scholar] [CrossRef]
- Kim, K.S.; Wang, J.W.; Kang, S.P. Tuning ionic liquids for hydrate inhibition. Chem. Comm. 2011, 47, 6341–6343. [Google Scholar] [CrossRef]
- Pham, T.P.T.; Cho, C.W.W.; Yun, Y.S.S.; Thuy Pham, T.P. Environmental fate and toxicity of ionic liquids: A review. Water Res. 2010, 44, 352–372. [Google Scholar] [CrossRef]
- Cha, J.H.; Seol, Y. Increasing Gas Hydrate Formation Temperature for Desalination of High Salinity Produced Water with Secondary Guests. ACS Sustain. Chem. Eng. 2013, 1, 1218–1224. [Google Scholar] [CrossRef]
- Madeira, P.P.; Bessa, A.; Alvares-Ribeiro, L.; Raquel Aires-Barros, M.; Rodrigues, A.E.; Uversky, V.N.; Zaslavsky, B.Y. Amino acid/water interaction study: A new amino acid scale. J. Biomol. Struct. Dyn. 2014, 32, 959–968. [Google Scholar] [CrossRef]
- Sa, J.H.; Lee, R.B.; Park, D.H.; Han, K.; Chun, H.D.; Lee, K.H. Amino acids as natural inhibitors for hydrate formation in CO2 sequestration. Environ. Sci. Technol. 2011, 45, 5885–5891. [Google Scholar] [CrossRef] [PubMed]
- Sa, J.H.; Kwak, G.H.; Han, K.; Ahn, D.; Cho, S.J.; Lee, J.D.; Lee, K.H. Inhibition of methane and natural gas hydrate formation by altering the structure of water with amino acids. Sci. Rep. 2016, 6, 31582. [Google Scholar] [CrossRef] [PubMed]
- Bavoh, C.B.; Lal, B.; Osei, H.; Sabil, K.M.; Mukhtar, H. A Review on the Role of Amino Acids in Gas Hydrate Inhibition, CO2 Capture and Sequestration, and natural Gas Storage. J. Nat. Gas Sci. Eng. 2019, 64, 52–71. [Google Scholar] [CrossRef]
- Badawy, W.A.; Ismail, K.M.; Fathi, A.M. Effect of Ni content on the corrosion behavior of Cu-Ni alloys in neutral chloride solutions. Electrochim. Acta 2005, 50, 3603–3608. [Google Scholar] [CrossRef]
- Nasir, Q.; Suleman, H.; Elsheikh, Y.A. A review on the role and impact of various additives as promoters/inhibitors for gas hydrate formation. J. Nat. Gas Sci. Eng. 2020, 76, 103211. [Google Scholar] [CrossRef]
- Laurent, S.; Forge, D.; Port, M.; Roch, A.; Robic, C.; Vander Elst, L.; Muller, R.N. Magnetic iron oxide nanoparticles: Synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications. Chem. Rev. 2008, 108, 2064–2110. [Google Scholar] [CrossRef]
- Tiwari, J.N.; Tiwari, R.N.; Kim, K.S. Zero-dimensional, one-dimensional, two-dimensional and three-dimensional nanostructured materials for advanced electrochemical energy devices. Prog. Mater. Sci. 2012, 57, 724–803. [Google Scholar] [CrossRef]
- Khan, I.; Saeed, K.; Khan, I. Nanoparticles: Properties, applications and toxicities. Arab. J. Chem. 2019, 12, 908–931. [Google Scholar] [CrossRef]
- Khan, M.S.; Bavoh, C.B.; Partoon, B.; Lal, B.; Bustam, M.A.; Shariff, A.M. Thermodynamic effect of ammonium based ionic liquids on CO2 hydrates phase boundary. J. Mol. Liq. 2017, 238, 533–539. [Google Scholar] [CrossRef]
- Gambelli, A.M.; Tinivella, U.; Giovannetti, R.; Castellani, B.; Giustiniani, M.; Rossi, A.; Zannotti, M.; Rossi, F. Observation of the Main Natural Parameters Influencing the Formation of Gas Hydrates. Energies 2021, 14, 1803. [Google Scholar] [CrossRef]
- Torres Pineda, I.; Lee, J.W.; Jung, I.; Kang, Y.T. CO2 absorption enhancement by methanol-based Al2O3 and SiO2 nanofluids in a tray column absorber. Int. J. Refrig. 2012, 35, 1402–1409. [Google Scholar] [CrossRef]
- Jung, J.Y.; Lee, J.W.; Kang, Y.T. CO2 absorption characteristics of nanoparticle suspension in methanol. J. Mech. Sci. Technol. 2012, 26, 2285–2290. [Google Scholar] [CrossRef]
- Yu, Y.S.; Zhou, S.D.; Li, X.S.; Wang, S.L. Effect of graphite nanoparticles on CO2 hydrate phase equilibrium. Fluid Phase Equilib. 2016, 414, 23–28. [Google Scholar] [CrossRef]
- Park, S.S.; Lee, S.B.; Kim, N.J. Effect of multi-walled carbon nanotubes on methane hydrate formation. J. Ind. Eng. Chem. 2010, 16, 551–555. [Google Scholar] [CrossRef]
- Mohammadi, M.; Haghtalab, A.; Fakhroueian, Z. Experimental study and thermodynamic modelling of CO2 gas hydrate formation in presence of zinc oxide nanoparticles. J. Chem. Therm. 2016, 96, 24–33. [Google Scholar] [CrossRef]
- Reichle, R.A.; McCurdy, K.G.; Hepler, L.G. Zinc hydroxide: Solubility product and hydroxy-complex stability constants from 12.5–75 °C. Can. J. Chem. 1975, 53, 3841–3845. [Google Scholar] [CrossRef]
- Li, J.; Liang, D.; Guo, K.; Wang, R.; Fan, S. Formation and dissociation of HFC134a gas hydrate in nano-copper suspension. Energy Convers. Manag. 2006, 47, 201–210. [Google Scholar] [CrossRef]
- Pahlavanzadeh, H.; Rezaei, S.; Khanlarkhani, M.; Manteghian, M.; Mohammadi, A.H. Kinetic study of methane hydrate formation in the presence of copper nanoparticles and CTAB. J. Nat. Gas Sci. Eng. 2016, 34, 803–810. [Google Scholar] [CrossRef]
- Arjang, S.; Manteghian, M.; Mohammadi, A. Effect of synthesized silver nanoparticles in promoting methane hydrate formation at 4.7 MPa and 5.7 MPa. Chem. Eng. Res. Des. 2013, 91, 1050–1054. [Google Scholar] [CrossRef]
- Mohammadi, A.; Manteghian, M.; Haghtalab, A.; Mohammadi, A.H.; Rahmati-Abkenar, M. Kinetic study of carbon dioxide hydrate formation in presence of silver nanoparticles and SDS. Chem. Eng. J. 2014, 237, 387–395. [Google Scholar] [CrossRef]
- Aliabadi, M.; Rasoolzadeh, A.; Esmaeilzadeh, F.; Alamdari, A. Experimental study of using CuO nanoparticles as a methane hydrate promoter. J. Nat. Gas Sci. Eng. 2015, 27, 1518–1522. [Google Scholar] [CrossRef]
- Najibi, H.; Mirzaee Shayegan, M.; Heidary, H. Experimental investigation of methane hydrate formation in the presence of copper oxide nanoparticles and SDS. J. Nat. Gas Sci. Eng. 2015, 23, 315–323. [Google Scholar] [CrossRef]
- Kedzierski, M.A.; Brignoli, R.; Quine, K.T.; Brown, J.S. Viscosity, density, and thermal conductivity of aluminum oxide and zinc oxide nanolubricants. Int. J. Refrig. 2017, 74, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Gambelli, A.M.; Stornelli, G.; Di Schino, A.; Rossi, F. Methane and Carbon Dioxide Hydrate Formation and Dissociation in Presence of a Pure Quartz Porous Framework Impregnated with CuSn12 Metallic powder: An Experimental Report. Materials 2021, 14, 5115. [Google Scholar] [CrossRef]
- Gambelli, A.M.; Stornelli, G.; Di Schino, A.; Rossi, F. Methane and carbon dioxide hydrates properties in presence of Inconel 718 particles: Analyses on its potential application in gas separation processes to perform efficiency improvement. J. Environ. Chem. Eng. 2021, 9, 106571. [Google Scholar] [CrossRef]
- Gambelli, A.M. Analyses on CH4 and CO2 hydrate formation to define the optimal pressure for CO2 injection to maximize the replacement efficiency into natural gas hydrate in presence of a silica-based natural porous medium, via depressurization techniques. Chem. Eng. Process. 2021, 167, 108512. [Google Scholar] [CrossRef]
- Gambelli, A.M.; Rossi, F. Experimental investigation on the possibility of defining the feasibility of CO2/CH4 exchange into a natural gas hydrate marine reservoir via fast analysis of sediment properties. Chem. Eng. Res. Des. 2021, 171, 327–339. [Google Scholar] [CrossRef]
- Wang, Y.; Feng, J.C.; Li, X.S.; Zhang, Y. Experimental investigation of optimization of well spacing for gas recovery from methane hydrate reservoir in sandy sediment by heat stimulation. Appl. Energy 2017, 207, 562–572. [Google Scholar] [CrossRef]
- Yuan, Q.; Sun, C.Y.; Yang, X.; Ma, P.C.; Ma, Z.W.; Liu, B.; Ma, Q.L.; Yang, L.Y.; Chen, G.J. Recovery of methane from hydrate reservoir with gaseous carbon dioxide using a three-dimensional middle-size reactor. Energy 2012, 40, 47–58. [Google Scholar] [CrossRef]
- Yin, Z.; Huang, L.; Linga, P. Effect of wellbore design on the production behaviour of methane hydrate-bearing sediments induced by depressurization. Appl. Energy 2019, 254, 113635. [Google Scholar] [CrossRef]
- Napoli, G.; Paura, M.; Vela, T.; Di Schino, A. Colouring titanium alloys by anodic oxidation. Metalurgija 2018, 57, 111–113. [Google Scholar]
- Stornelli, G.; Ridolfi, M.; Folgarait, P.; De Nisi, J.; Corapi, D.; Repitsch, C.; Di Schino, A. Feasibility assessment of magnetic cores through additive manufacturing techniques. Metall. Ital. 2021, 113, 50–61. [Google Scholar]
- Stornelli, G.; Gaggia, G.; Rallini, M.; Di Schino, A. Heat treatment effect on managing steel manufactured by laser powder bed fusion technology: Microstructure and mechanical properties. Acta Metall. Slovaca 2021, 27, 122–126. [Google Scholar] [CrossRef]
- Gambelli, A.M. An experimental description of the double positive effect of CO2 injection in methane hydrate deposits in terms of climate change mitigation. Chem. Eng. Sci. 2021, 233, 116430. [Google Scholar] [CrossRef]
- Gambelli, A.M.; Filipponi, M.; Rossi, F. How methane release may affect carbon dioxide storage during replacement processes in natural gas hydrate reservoirs. J. Pet. Sci. Eng. 2021, 205, 108895. [Google Scholar] [CrossRef]
- Nema, Y.; Ohmura, R.; Senaha, I.; Yasuda, K. Quadruple point determination in carbon dioxide hydrate forming system. Fluid Phase Equilib. 2017, 441, 49–53. [Google Scholar] [CrossRef]
- Nagashima, H.D.; Fukushima, N.; Ohmura, R. Phase equilibrium condition measurements in carbon dioxide clathrate hydrate forming system from 199.1 K to 247.1 K. Fluid Phase Equilib. 2016, 413, 53–56. [Google Scholar] [CrossRef]
- Kassim, Z.; Khan, M.S.; Lal, B. Thermodynamic modelling on methane hydrate equilibrium condition in the presence of electrolyte inhibitor. Mater. Today 2019, 19, 1395–1402. [Google Scholar] [CrossRef]
- Rossi, F.; Gambelli, A.M.; Sharma, D.K.; Castellani, B.; Nicolini, A.; Castaldi, M.J. Experiments on methane hydrates formation in seabed deposits and gas recovery adopting carbon dioxide replacement strategies. Appl. Therm. Eng. 2019, 148, 371–381. [Google Scholar] [CrossRef]
- Takeya, S.; Kida, M.; Minami, H.; Sakagami, H.; Hachikubo, A.; Takahashi, N.; Shoji, H.; Soloviev, V.; Wallmann, K.; Biebow, N.; et al. Structure and thermal expansion of natural gas clathrate hydrates. Chem. Eng. Sci. 2006, 61, 2670–2674. [Google Scholar] [CrossRef]
- Aregba, A.G. Gas Hydrate—Properties, Formation and Benefits. Open J. Yangtze Gas Oil 2017, 2, 27–44. [Google Scholar] [CrossRef] [Green Version]
- Papadimitriou, N.I.; Tsimpanogiannis, I.N.; Economou, I.G.; Studos, A.K. Storage of Methane in Clathrate Hydrates: Monte Carlo Simulations of sI Hydrates and Comparison with Experimental Measurements. J. Chem. Eng. Data 2016, 61, 2886–2896. [Google Scholar] [CrossRef]
- Fitzgerald, G.C.; Castaldi, M.J.; Zhou, Y. Large scale reactor details and results for the formation and decomposition of methane hydrates via thermal stimulation dissociation. J. Pet. Sci. Eng. 2012, 94–95, 19–27. [Google Scholar] [CrossRef]
- Castellani, B.; Rossetti, G.; Tupsakhare, S.; Rossi, F.; Nicolini, A.; Castaldi, M.J. Simulation of CO2 storage and methane gas production from gas hydrates in a large scale laboratory reactor. J. Pet. Sci. Eng. 2016, 147, 515–527. [Google Scholar] [CrossRef]
- Sun, J.; Zin, Y.; Chou, I.M.; Sun, R.; Jiang, L. Hydrate Stability in the H2S-H2O system Visual Observations and Measuremnts in a High-Pressure Optical Cell and Thermodynamic Models. J. Chem. Eng. Data 2020, 65, 3884–3892. [Google Scholar] [CrossRef]
- Chou, I.-M.; Song, Y.; Burruss, R.C. A new method for synthesizing fluid inclusions in fused silica capillaries containing organic and inorganic material. Geochim. Cosmochim. Acta 2008, 72, 5217–5231. [Google Scholar] [CrossRef]
- Kvamme, B. Kinetics of hydrate formation, dissociation and reformation. Chem. Thermodyn. Therm. Anal. 2021, 1–2, 100004. [Google Scholar] [CrossRef]
- Kvamme, B.; Zhao, J.Z.; Wei, N.; Sun, W.T.; Saeidi, N.; Pei, J.; Kuznetsova, T. Hydrate production philosophy and thermodynamic calculations. Energies 2020, 13, 672. [Google Scholar] [CrossRef] [Green Version]
- Kvamme, B. Kinetics of hydrate formation from nucleation theory. Int. J. Offshore Polar Eng. 2002, 12, 256–263. [Google Scholar]
- Kvamme, B. Droplets of dry ice and cold liquid CO2 for self-transport of CO2 to large depths. Int. J. Offshore Polar Eng. 2003, 13, 139–146. [Google Scholar]
- Rossi, F.; Li, Y.; Gambelli, A.M. Thermodynamic and Kinetic Description of the Main Effects Related to the Memory Effect during Carbon Dioxide Hydrates Formation in a Confined Environment. Sustainability 2021, 13, 13797. [Google Scholar] [CrossRef]
- Kashchiev, D.; Verdoes, D.; van Rosmalen, G.M. Induction time and metastability limit in new phase formation. J. Cryst. Growth 1991, 110, 373–380. [Google Scholar] [CrossRef]
- Wang, S.R.; Yang, M.J.; Liu, W.G.; Zhao, J.F.; Song, Y.C. Investigation on the induction time of methane hydrate formation in porous media under quiescent conditions. J. Pet. Sci. Eng. 2016, 145, 565–572. [Google Scholar] [CrossRef]
- Englezos, P.; Kalogerakis, N.; Dholabai, P.D.; Bishnoi, P.R. Kinetics of formation of methane and ethane gas hydrates. Chem. Eng. Sci. 1987, 42, 2647–2658. [Google Scholar] [CrossRef]
- Gambelli, A.M.; Rossi, F. Formation rate as parameter to distinguish nucleation from hydrate massive growth phase: Experimental investigation in presence of two different porous media. Exp. Therm. Fluid Sci. 2022, 131, 110525. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ti23 8.68 [wt%] | Ti23 26.04 [wt%] | Ti23 8.68 [wt%] | Ti23 26.04 [wt%] | ||
|---|---|---|---|---|---|
| Temperature [°C] | Pressure [bar] | Pressure [bar] | Temperature [°C] | Pressure [bar] | Pressure [bar] |
| 0.0 | - | 29.08 | 3.5 | 38.46 | 38.64 |
| 0.1 | - | 29.49 | 3.6 | 38.86 | 38.96 |
| 0.2 | - | 29.83 | 3.7 | 39.26 | 39.3 |
| 0.3 | - | 29.98 | 3.8 | 39.52 | 39.6 |
| 0.4 | - | 30.18 | 3.9 | 39.93 | 40 |
| 0.5 | - | 30.42 | 4.0 | 40.32 | 40.25 |
| 0.6 | - | 30.6 | 4.1 | 40.6 | 40.51 |
| 0.7 | - | 30.93 | 4.2 | 41 | 40.92 |
| 0.8 | - | 31.21 | 4.3 | 41.33 | 41.26 |
| 0.9 | - | 31.43 | 4.4 | 41.74 | 41.49 |
| 1.0 | 30.59 | 31.63 | 4.5 | 42.08 | 41.82 |
| 1.1 | 30.92 | 32.02 | 4.6 | 42.41 | 42.08 |
| 1.2 | 31.2 | 32.18 | 4.7 | 42.8 | 42.58 |
| 1.3 | 31.43 | 32.43 | 4.8 | 43.07 | 42.99 |
| 1.4 | 31.62 | 32.7 | 4.9 | 43.49 | 43.21 |
| 1.5 | 31.99 | 32.91 | 5.0 | 43.76 | 43.49 |
| 1.6 | 32.18 | 33.08 | 5.1 | 43.89 | 43.81 |
| 1.7 | 32.59 | 33.32 | 5.2 | 44.08 | 44 |
| 1.8 | 32.83 | 33.48 | 5.3 | 44.3 | 44.3 |
| 1.9 | 33.18 | 33.67 | 5.4 | 44.41 | 44.49 |
| 2.0 | 33.39 | 33.89 | 5.5 | 44.57 | 44.7 |
| 2.1 | 33.8 | 34 | 5.6 | 44.81 | 44.98 |
| 2.2 | 34.08 | 39.17 | 5.7 | 44.9 | 45.22 |
| 2.3 | 34.41 | 34.58 | 5.8 | 45.09 | 45.38 |
| 2.4 | 34.83 | 34.91 | 5.9 | 45.3 | 45.57 |
| 2.5 | 34.99 | 35.17 | 6.0 | 45.49 | 45.78 |
| 2.6 | 35.39 | 35.39 | 6.1 | 45.57 | 45.97 |
| 2.7 | 35.81 | 35.8 | 6.2 | 45.79 | 46.19 |
| 2.8 | 36.08 | 36.17 | 6.3 | 45.98 | 46.28 |
| 2.9 | 36.5 | 36.58 | 6.4 | 46.06 | 46.39 |
| 3.0 | 36.83 | 36.9 | 6.5 | 46.28 | 46.55 |
| 3.1 | 37.09 | 37.31 | 6.6 | 46.39 | 46.69 |
| 3.2 | 37.5 | 37.51 | 6.7 | 46.56 | 46.8 |
| 3.3 | 37.9 | 37.98 | 6.8 | -- | 46.83 |
| 3.4 | 38.17 | 38.3 | 6.9 | -- | 46.88 |
| Ti23 8.68 [wt%] | Ti23 26.04 [wt%] | Ti23 8.68 [wt%] | Ti23 26.04 [wt%] | ||
|---|---|---|---|---|---|
| Temperature [°C] | Pressure [bar] | Pressure [bar] | Temperature [°C] | Pressure [bar] | Pressure [bar] |
| 0.0 | - | 13.9 | 5.0 | 23.9 | 24.31 |
| 0.1 | - | 13.98 | 5.1 | 24.39 | 24.48 |
| 0.2 | - | 14.09 | 5.2 | 24.59 | 24.79 |
| 0.3 | - | 14.17 | 5.3 | 24.79 | 25.15 |
| 0.4 | - | 14.3 | 5.4 | 25.06 | 25.48 |
| 0.5 | - | 14.38 | 5.5 | 25.28 | 25.89 |
| 0.6 | - | 14.57 | 5.6 | 25.4 | 26.08 |
| 0.7 | - | 14.65 | 5.7 | 25.56 | 26.46 |
| 0.8 | - | 14.89 | 5.8 | 25.67 | 26.79 |
| 0.9 | - | 14.97 | 5.9 | 25.89 | 27.07 |
| 1.0 | 15.86 | 15.16 | 6.0 | 25.97 | 27.57 |
| 1.1 | 15.97 | 15.3 | 6.1 | 26.08 | 27.9 |
| 1.2 | 15.99 | 15.46 | 6.2 | 26.3 | 28.31 |
| 1.3 | 16.13 | 15.57 | 6.3 | 26.35 | 28.5 |
| 1.4 | 16.17 | 15.78 | 6.4 | 26.42 | 29 |
| 1.5 | 16.35 | 15.86 | 6.5 | 26.57 | 29.42 |
| 1.6 | 16.55 | 16.05 | 6.6 | 26.65 | 29.91 |
| 1.7 | 16.77 | 16.27 | 6.7 | 26.79 | 30.27 |
| 1.8 | 16.88 | 16.46 | 6.8 | 27.19 | 30.6 |
| 1.9 | 17.04 | 16.63 | 6.9 | 27.83 | 31.09 |
| 2.0 | 17.15 | 16.88 | 7.0 | 28.26 | 31.52 |
| 2.1 | 17.37 | 16.96 | 7.1 | 28.81 | 31.91 |
| 2.2 | 17.46 | 17.29 | 7.2 | 29.33 | 32.32 |
| 2.3 | 17.64 | 17.37 | 7.3 | 29.7 | 32.7 |
| 2.4 | 17.78 | 17.64 | 7.4 | 30.19 | 33.19 |
| 2.5 | 18.05 | 17.85 | 7.5 | 30.62 | 33.67 |
| 2.6 | 18.27 | 18.04 | 7.6 | 31.07 | 34.09 |
| 2.7 | 18.46 | 18.13 | 7.7 | 31.41 | 34.59 |
| 2.8 | 18.65 | 18.46 | 7.8 | 31.83 | 35.11 |
| 2.9 | 18.87 | 18.65 | 7.9 | 32.09 | 35.51 |
| 3.0 | 19.06 | 18.87 | 8.0 | 32.57 | 36.09 |
| 3.1 | 19.35 | 19.14 | 8.1 | 32.86 | 36.58 |
| 3.2 | 19.55 | 19.35 | 8.2 | 33.16 | 37.1 |
| 3.3 | 19.77 | 19.63 | 8.3 | 33.4 | 37.66 |
| 3.4 | 19.97 | 19.86 | 8.4 | 33.71 | 38.31 |
| 3.5 | 20.28 | 20.05 | 8.5 | 34.17 | 38.66 |
| 3.6 | 20.47 | 20.28 | 8.6 | 34.46 | 39.37 |
| 3.7 | 20.81 | 20.56 | 8.7 | 34.58 | 39.74 |
| 3.8 | 20.89 | 20.89 | 8.8 | 34.7 | 40.41 |
| 3.9 | 21.16 | 21.09 | 8.9 | - | 40.82 |
| 4.0 | 21.49 | 21.38 | 9.0 | - | 41.34 |
| 4.1 | 21.66 | 21.58 | 9.1 | - | 41.74 |
| 4.2 | 21.99 | 21.91 | 9.2 | - | 42.09 |
| 4.3 | 22.18 | 22.18 | 9.3 | - | 42.42 |
| 4.4 | 22.48 | 22.41 | 9.4 | - | 42.81 |
| 4.5 | 22.68 | 22.82 | 9.5 | - | 43.08 |
| 4.6 | 22.89 | 22.97 | 9.6 | - | 43.41 |
| 4.7 | 23.17 | 23.3 | 9.7 | - | 43.6 |
| 4.8 | 23.49 | 23.66 | 9.8 | - | 43.72 |
| 4.9 | 23.66 | 23.91 | 9.9 | - | 43.83 |
| Test n° | Guest | Ti23 [wt%] | Induction Time [h] |
|---|---|---|---|
| A | CH4 | 8.68 | 2.75 |
| B | CH4 | 8.68 | 6.66 |
| C | CO2 | 8.68 | 0.82 |
| D | CO2 | 8.68 | 0.9 |
| E | CH4 | 26.04 | 2.93 |
| F | CH4 | 26.04 | 1.38 |
| G | CO2 | 26.04 | 0.61 |
| H | CO2 | 26.04 | 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambelli, A.M.; Stornelli, G.; Di Schino, A.; Rossi, F. Formation and Dissociation of CH4 and CO2 Hydrates in Presence of a Sediment Composed by Pure Quartz Mixed with Ti23 Particles. Materials 2022, 15, 1470. https://doi.org/10.3390/ma15041470
Gambelli AM, Stornelli G, Di Schino A, Rossi F. Formation and Dissociation of CH4 and CO2 Hydrates in Presence of a Sediment Composed by Pure Quartz Mixed with Ti23 Particles. Materials. 2022; 15(4):1470. https://doi.org/10.3390/ma15041470
Chicago/Turabian StyleGambelli, Alberto Maria, Giulia Stornelli, Andrea Di Schino, and Federico Rossi. 2022. "Formation and Dissociation of CH4 and CO2 Hydrates in Presence of a Sediment Composed by Pure Quartz Mixed with Ti23 Particles" Materials 15, no. 4: 1470. https://doi.org/10.3390/ma15041470