
Electronic and Optical Properties of Twin T-Graphene Co-Doped with Boron and Phosphorus


Abstract
:1. Introduction
2. Methodology
3. Results and Discussion
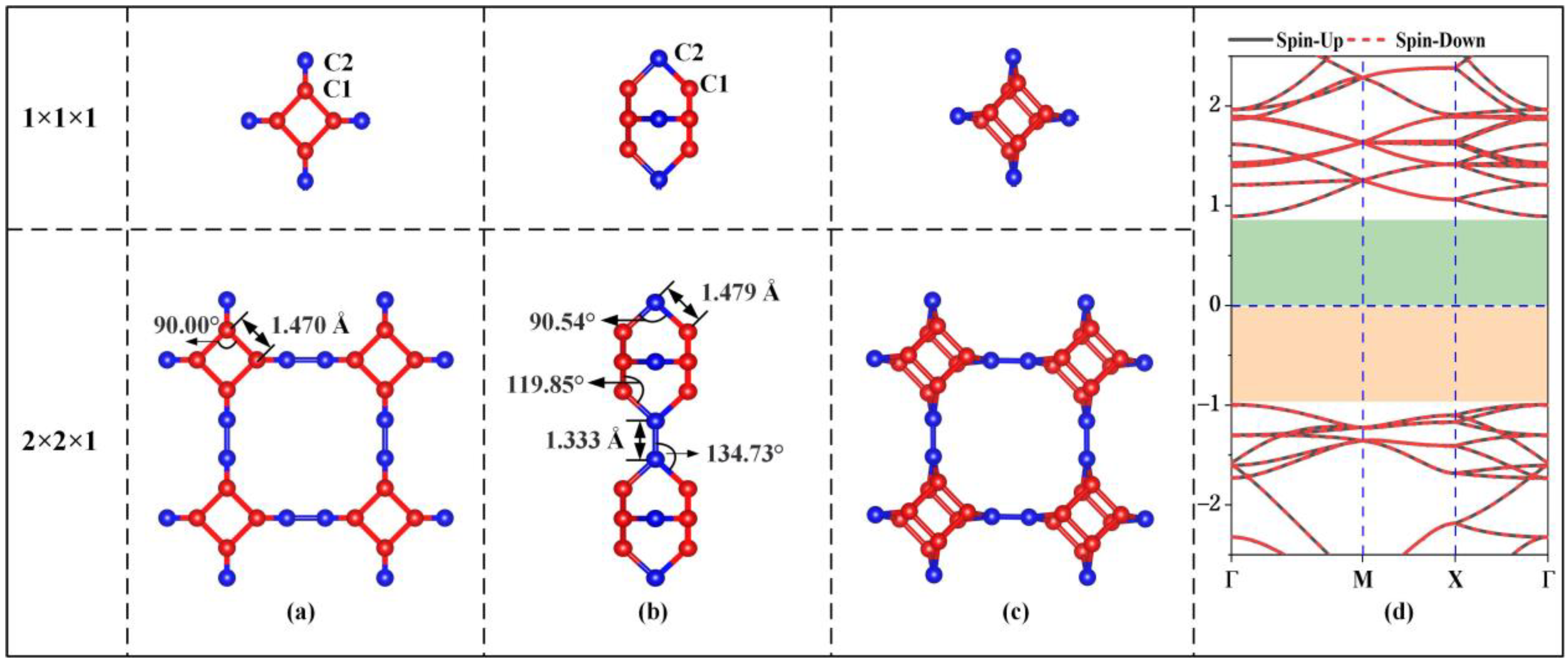
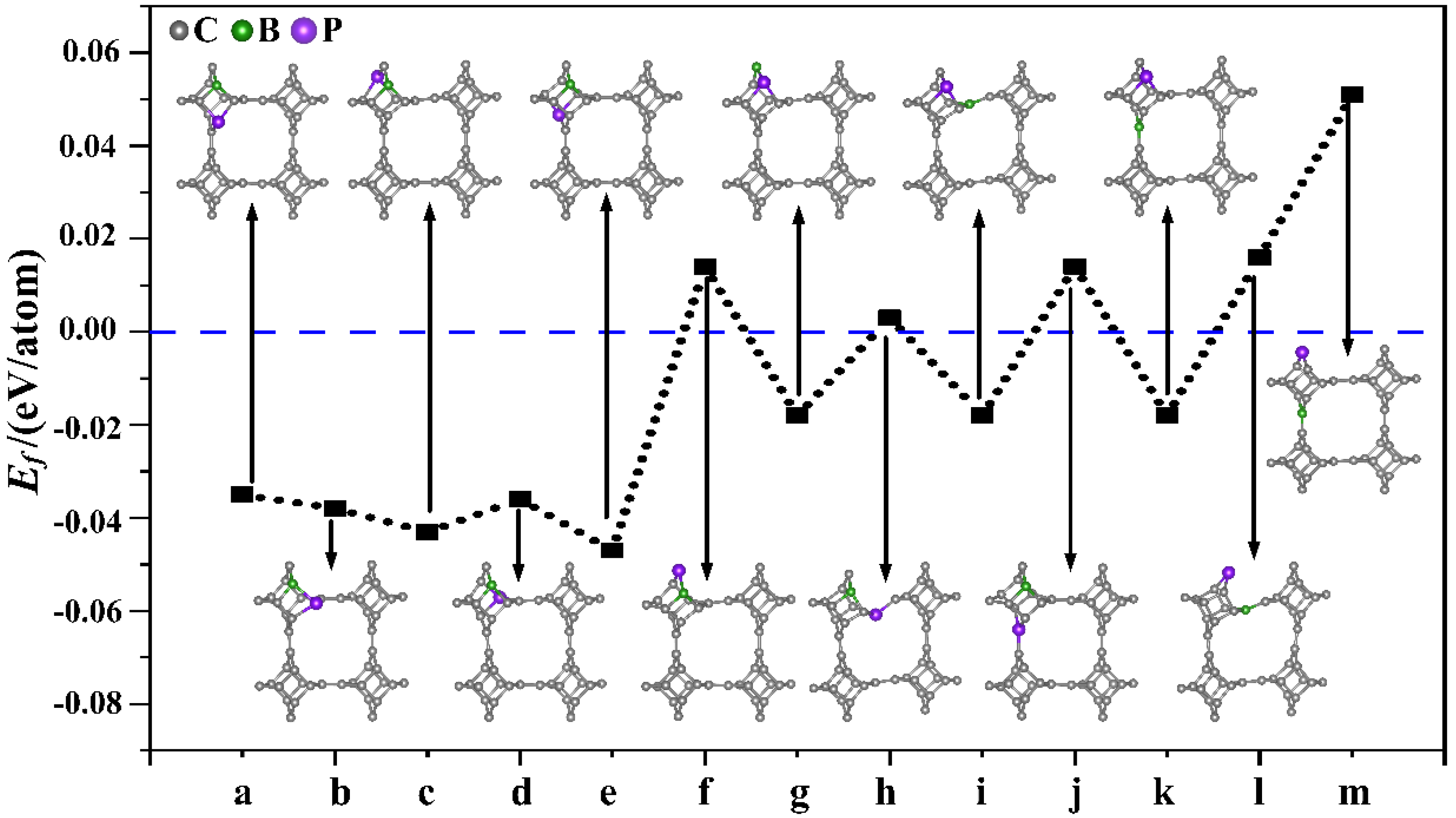
3.1. Structural Stability
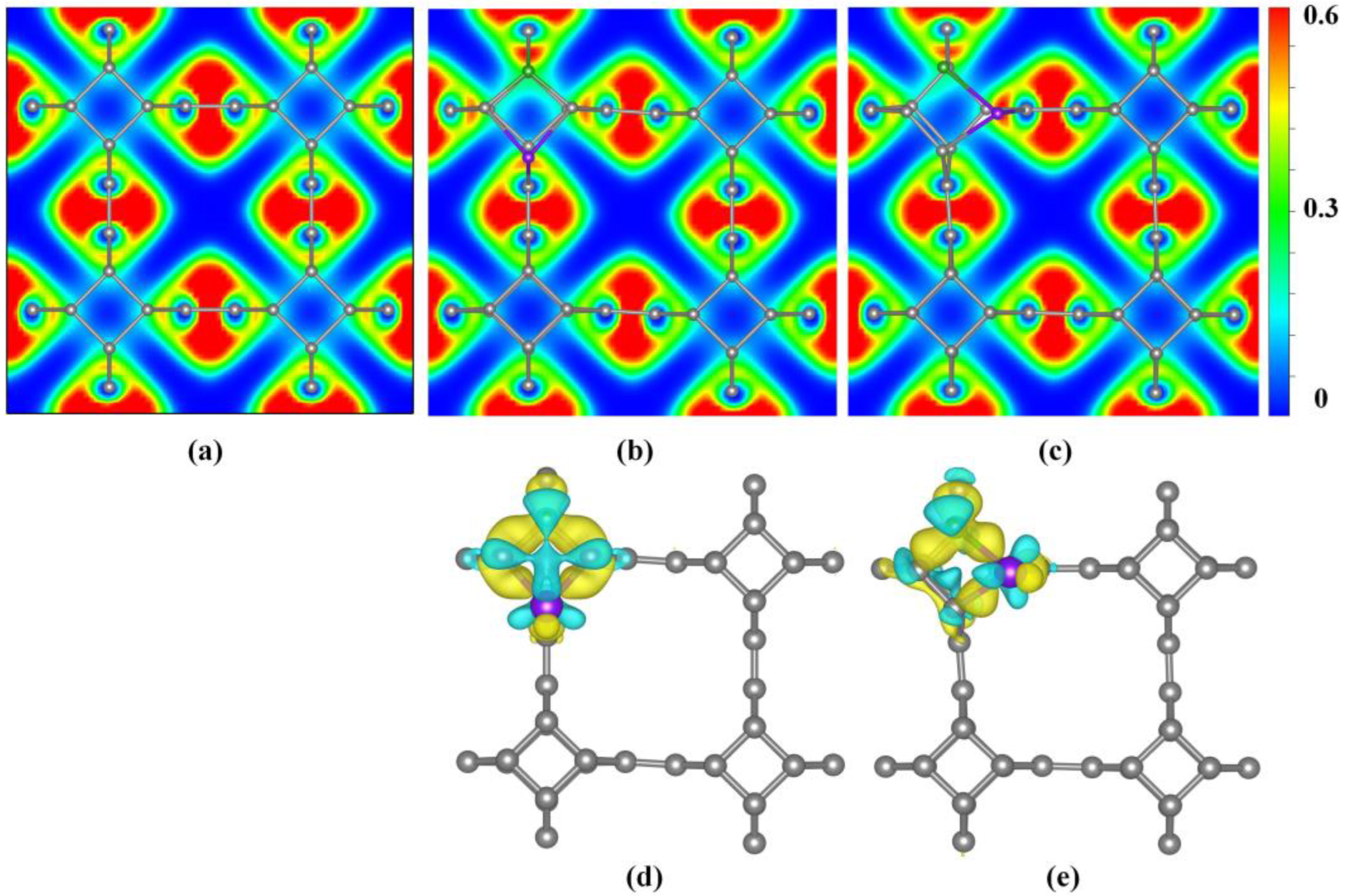
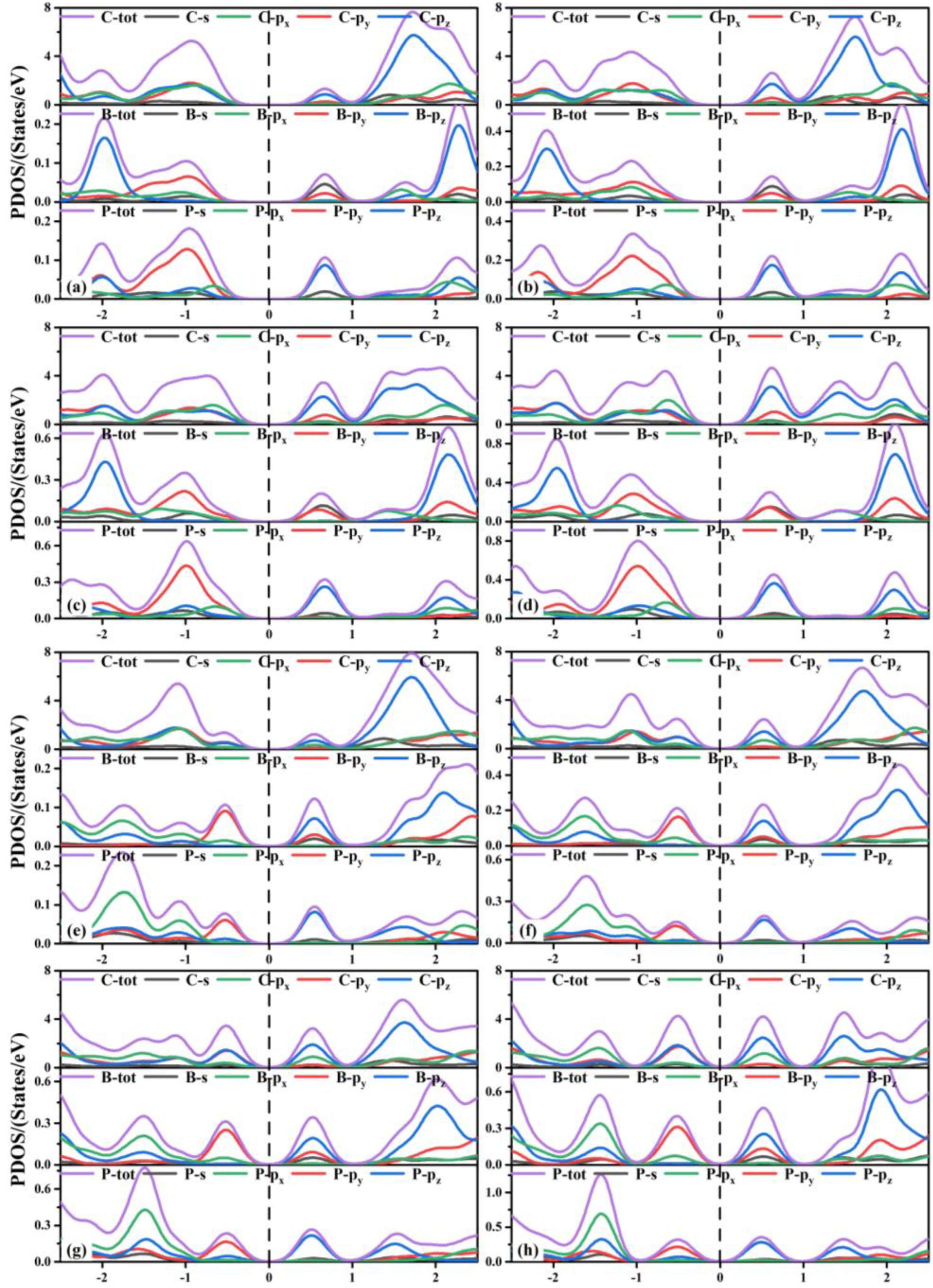
3.2. Electronic Properties
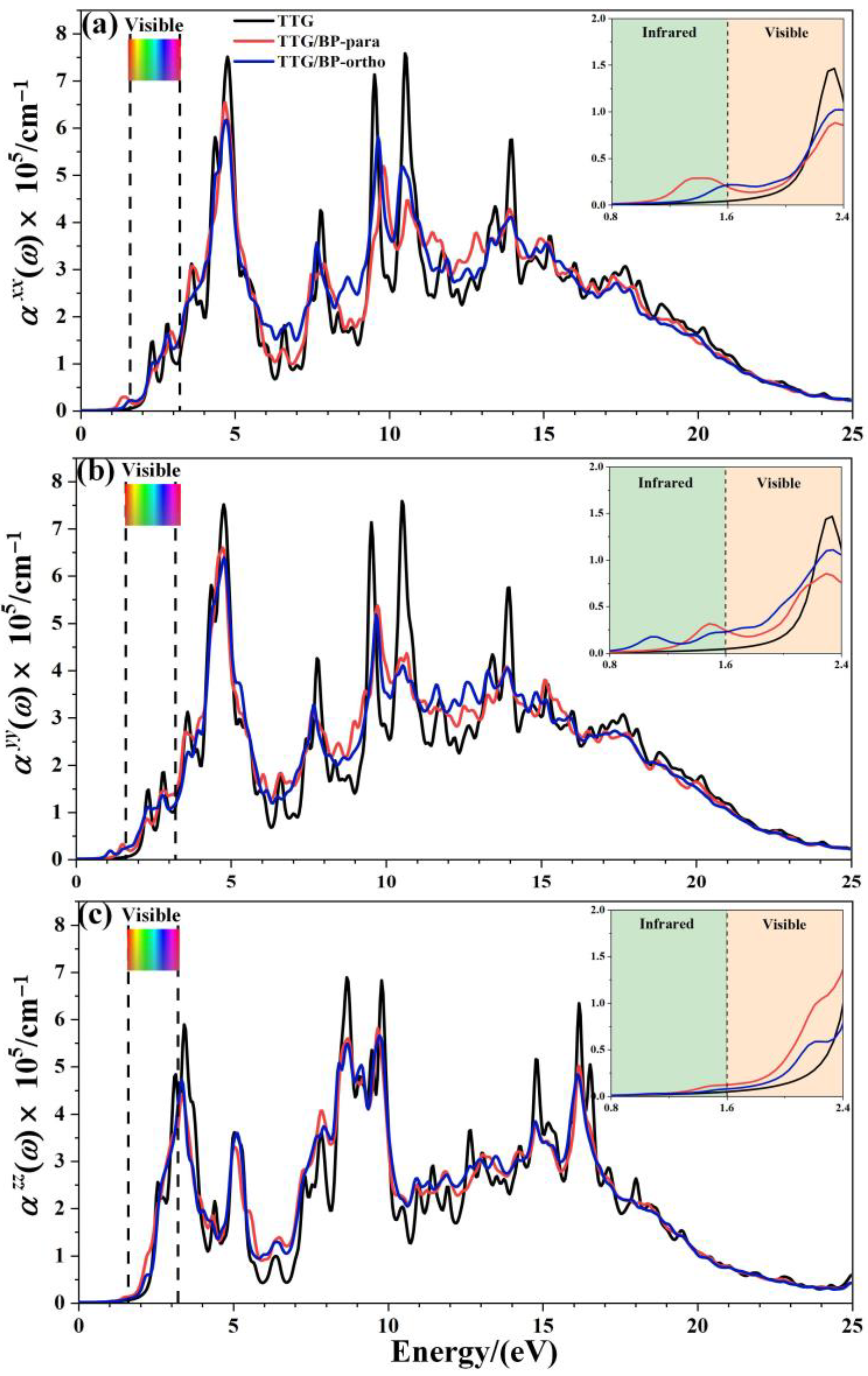
3.3. Optical Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bhattacharya, D.; Jana, D. Twin T-graphene: A new semiconducting 2D carbon allotrope. Phys. Chem. Chem. Phys. 2020, 22, 10286–10294. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.W.; Leng, J.; Li, J.; Guo, Z.; Chang, T.; Guo, X.; Zhang, T. Twin graphene: A novel two-dimensional semiconducting carbon allotrope. Carbon 2017, 118, 370–375. [Google Scholar] [CrossRef]
- Yu, B.-Y.; Xie, Y.; Wu, X.; Gao, Y.; Wang, S.-F.; Zhang, J.-M. Structural and electronic properties of AlY (Y B, N, O) dual-doped twin graphene: A density functional theory study. Phys. E Low-Dimens. Syst. Nanostruct. 2021, 128, 114619. [Google Scholar] [CrossRef]
- Majidi, R.; Ramazani, A.; Rabczuk, T. Electronic properties of transition metal embedded twin T-graphene: A density functional theory study. Phys. E Low-Dimens. Syst. Nanostruct. 2021, 133, 114806. [Google Scholar] [CrossRef]
- Li, L.; Zhang, H.; Cheng, X.; Miyamoto, Y. First-principles studies on 3d transition metal atom adsorbed twin graphene. Appl. Surf. Sci. 2018, 441, 647–653. [Google Scholar] [CrossRef]
- Majidi, R.; Ramazani, A. Detection of HF and H2S with pristine and Ti-embedded twin graphene: A density functional theory study. J. Phys. Chem. Solids 2019, 132, 31–37. [Google Scholar] [CrossRef]
- Majidi, R.; Nadafan, M. Detection of exhaled gas by γ-graphyne and twin-graphene for early diagnosis of lung cancer: A density functional theory study. Phys. Lett. A 2020, 384, 126036. [Google Scholar] [CrossRef]
- Majidi, R.; Rabczuk, T. Structural and electronic properties of BN co-doped and BN analogue of twin graphene sheets: A density functional theory study. J. Phys. Chem. Solids 2019, 135, 109115. [Google Scholar] [CrossRef]
- Montejo-Alvaro, F.; Gonzalez-Quijano, D.; Valmont-Pineda, J.A.; Rojas-Chavez, H.; Juarez-Garcia, J.M.; Medina, D.I.; Cruz-Martinez, H. CO2 adsorption on PtCu sub-nanoclusters deposited on pyridinic N-doped graphene: A DFT investigation. Materials 2021, 14, 7619. [Google Scholar] [CrossRef]
- Ren, F.; Yao, M.L.; Li, M.; Wang, H. Tailoring the structural and electronic properties of graphene through ion implantation. Materials 2021, 14, 5080. [Google Scholar] [CrossRef]
- Armaković, S.; Armaković, S.J. Investigation of boron modified graphene nanostructures; optoelectronic properties of graphene nanoparticles and transport properties of graphene nanosheets. J. Phys. Chem. Solids 2016, 98, 156–166. [Google Scholar] [CrossRef]
- Xie, Y.; Cao, S.; Wu, X.; Yu, B.-Y.; Chen, L.-Y.; Zhang, J.-M. Density functional theory study of hydrogen sulfide adsorption onto transition metal-doped bilayer graphene using external electric fields. Phys. E Low-Dimens. Syst. Nanostruct. 2020, 124, 114252. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, W.; Cao, S.; Zhou, A.; Zhang, J. First-principles study of transition metal monatomic chains intercalated AA-stacked bilayer graphene nanoribbons. Phys. E Low-Dimens. Syst. Nanostruct. 2019, 106, 114–120. [Google Scholar] [CrossRef]
- Choi, W.I.; Jhi, S.-H.; Kim, K.; Kim, Y.-H. Divacancy-nitrogen-assisted transition metal dispersion and hydrogen adsorption in defective graphene: A first-principles study. Phys. Rev. B 2010, 81, 085441. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; He, W.; Wang, X.; Wang, X.; Song, M.; Zhao, J. N/S dual-doped graphene with high defect density for enhanced supercapacitor properties. Int. J. Hydrogen Energy 2020, 45, 112–122. [Google Scholar] [CrossRef]
- Denis, P.A.; Iribarne, F. The effect of the dopant nature on the reactivity, interlayer bonding and electronic properties of dual doped bilayer graphene. Phys. Chem. Chem. Phys. 2016, 18, 24693–24703. [Google Scholar] [CrossRef]
- Denis, P.A.; Pereyra Huelmo, C. Structural characterization and chemical reactivity of dual doped graphene. Carbon 2015, 87, 106–115. [Google Scholar] [CrossRef]
- Denis, P.A.; Huelmo, C.P.; Martins, A.S. Band gap opening in dual-doped monolayer graphene. J. Phys. Chem. C 2016, 120, 7103–7112. [Google Scholar] [CrossRef]
- Denis, P.A. Mono and dual doped monolayer graphene with aluminum, silicon, phosphorus and sulfur. Comput. Theor. Chem. 2016, 1097, 40–47. [Google Scholar] [CrossRef]
- Denis, P.A.; Huelmo, C.P.; Iribarne, F. Theoretical characterization of sulfur and nitrogen dual-doped graphene. Comput. Theor. Chem. 2014, 1049, 13–19. [Google Scholar] [CrossRef]
- Deb, J.; Sarkar, U. Boron-nitride and boron-phosphide doped twin-graphene: Applications in electronics and optoelectronics. Appl. Surf. Sci. 2021, 541, 148657. [Google Scholar] [CrossRef]
- Zhang, L.; Liang, P.; Man, X.; Wang, D.; Huang, J.; Shu, H.; Liu, Z.; Wang, L. Fe, N co-doped graphene as a multi-functional anchor material for lithium-sulfur battery. J. Phys. Chem. Solids 2019, 126, 280–286. [Google Scholar] [CrossRef]
- Chae, G.S.; Youn, D.H.; Lee, J.S. Nanostructured iron sulfide/N, S dual-doped carbon nanotube-graphene composites as efficient electrocatalysts for oxygen reduction reaction. Materials 2021, 14, 2146. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, S.S.; Kim, H.S.; Kim, Y.H. Anomalous transport properties in boron and phosphorus co-doped armchair graphene nanoribbons. Nanotechnology 2016, 27, 47LT01. [Google Scholar] [CrossRef]
- Denis, P.A. Lithium adsorption on heteroatom mono and dual doped graphene. Chem. Phys. Lett. 2017, 672, 70–79. [Google Scholar] [CrossRef]
- Wang, D.; Liu, T.; Wang, J.; Wu, Z. N, P (S) Co-doped Mo2C/C hybrid electrocatalysts for improved hydrogen generation. Carbon 2018, 139, 845–852. [Google Scholar] [CrossRef]
- Li, Z.; Yang, H.; Zhang, D.; Zhou, W.; Gao, N.; Wang, J.; Yang, D. Effects of Bi and S co-doping on the enhanced photoelectric performance of ZnO: Theoretical and experimental investigations. J. Alloys Compd. 2021, 872, 159648. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Gong, Z.; Xie, J.; Lu, T.; Pan, L. Nitrogen and sulfur co-doped vanadium carbide MXene for highly reversible lithium-ion storage. J. Colloid Interface Sci. 2021, 587, 489–498. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, N.; Yao, H.; Liu, D.; Song, L.; Zhu, J.; Li, S.; Gu, L.; Lin, K.; Wang, D. Stereo-defined Codoping of sp-N and S Atoms in Few-Layer Graphdiyne for Oxygen Evolution Reaction. J. Am. Chem. Soc. 2019, 141, 7240–7244. [Google Scholar] [CrossRef]
- Liang, G.; Wu, Z.; Didier, C.; Zhang, W.; Cuan, J.; Li, B.; Ko, K.Y.; Hung, P.Y.; Lu, C.Z.; Chen, Y.; et al. A Long Cycle-Life High-Voltage Spinel Lithium-Ion Battery Electrode Achieved by Site-Selective Doping. Angew. Chem. Int. Ed. Engl. 2020, 59, 10594–10602. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B Condens. Matter 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bučko, T.; Hafner, J.; Lebègue, S.; Ángyán, J.G. Improved description of the structure of molecular and layered crystals: Ab initio DFT calculations with van der Waals corrections. J. Phys. Chem. A 2010, 114, 11814–11824. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B Condens. Matter 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Sanville, E.; Kenny, S.D.; Smith, R.; Henkelman, G. Improved grid-based algorithm for Bader charge allocation. J. Comput. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef]
- Qiu, B.; Zhao, X.; Hu, G.; Yue, W.; Ren, J.; Yuan, X. Optical properties of graphene/MoS2 heterostructure: First principles calculations. Nanomaterials 2018, 8, 962. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Deslippe, J.; Park, C.; Cohen, M.; Louie, S. Excitonic effects on the optical response of graphene and bilayer graphene. Phys. Rev. Lett. 2009, 103, 186802. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Concentration (%) | Ef (eV) | Band Gap (eV) |
|---|---|---|---|
| Para | 4.2 | −0.035 | 1.28 |
| 8.3 | −0.069 | 1.12 | |
| 12.5 | −0.100 | 1.00 | |
| 16.7 | −0.131 | 0.98 | |
| Ortho | 4.2 | −0.038 | 1.04 |
| 8.3 | −0.075 | 0.86 | |
| 12.5 | –0.112 | 0.80 | |
| 16.7 | −0.148 | 0.70 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Xie, Y.; Wang, S.; Li, S.; Chen, L.; Zhang, J. Electronic and Optical Properties of Twin T-Graphene Co-Doped with Boron and Phosphorus. Materials 2022, 15, 2876. https://doi.org/10.3390/ma15082876
Gao Y, Xie Y, Wang S, Li S, Chen L, Zhang J. Electronic and Optical Properties of Twin T-Graphene Co-Doped with Boron and Phosphorus. Materials. 2022; 15(8):2876. https://doi.org/10.3390/ma15082876
Chicago/Turabian StyleGao, Yue, You Xie, Sufang Wang, Shuang Li, Liyong Chen, and Jianmin Zhang. 2022. "Electronic and Optical Properties of Twin T-Graphene Co-Doped with Boron and Phosphorus" Materials 15, no. 8: 2876. https://doi.org/10.3390/ma15082876
APA StyleGao, Y., Xie, Y., Wang, S., Li, S., Chen, L., & Zhang, J. (2022). Electronic and Optical Properties of Twin T-Graphene Co-Doped with Boron and Phosphorus. Materials, 15(8), 2876. https://doi.org/10.3390/ma15082876