
Stereoselective Synthesis and Anticancer Activity of 2,6-Disubstituted trans-3-Methylidenetetrahydropyran-4-ones

 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.2. Biology
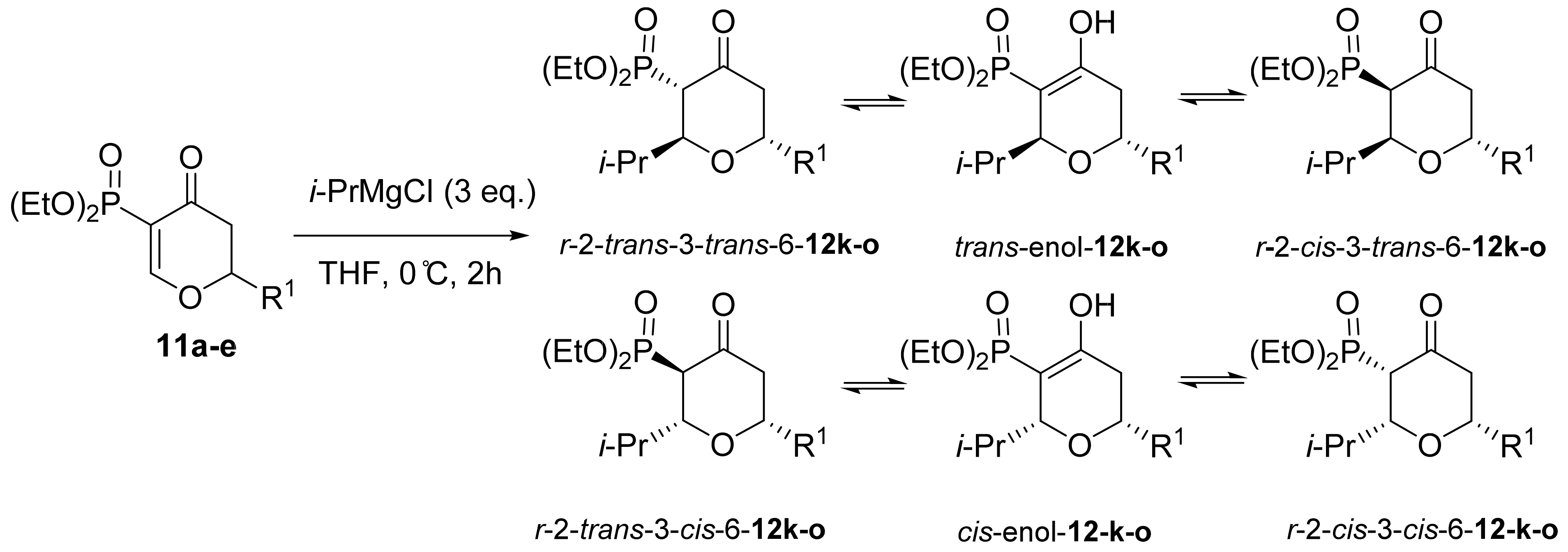
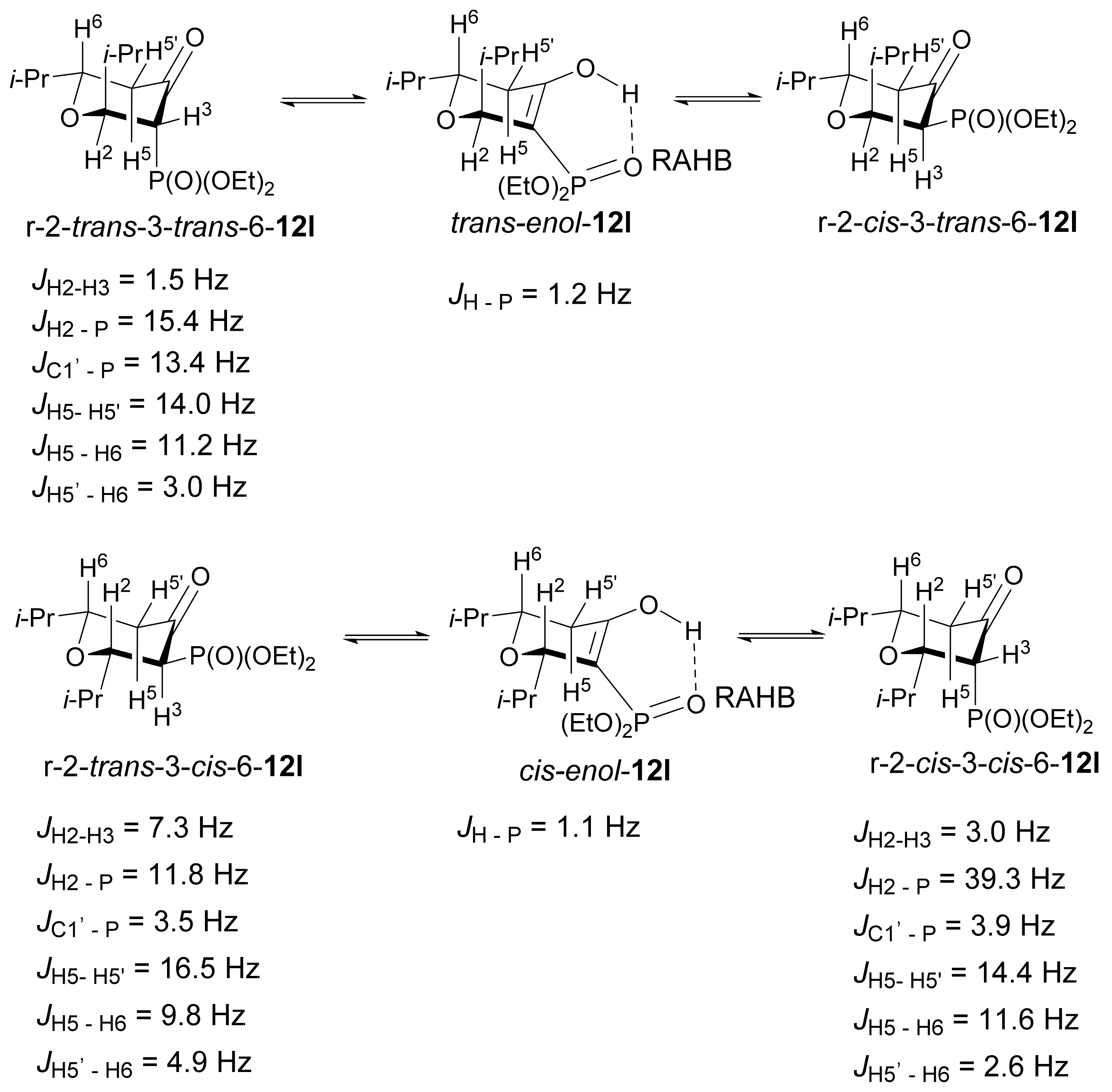
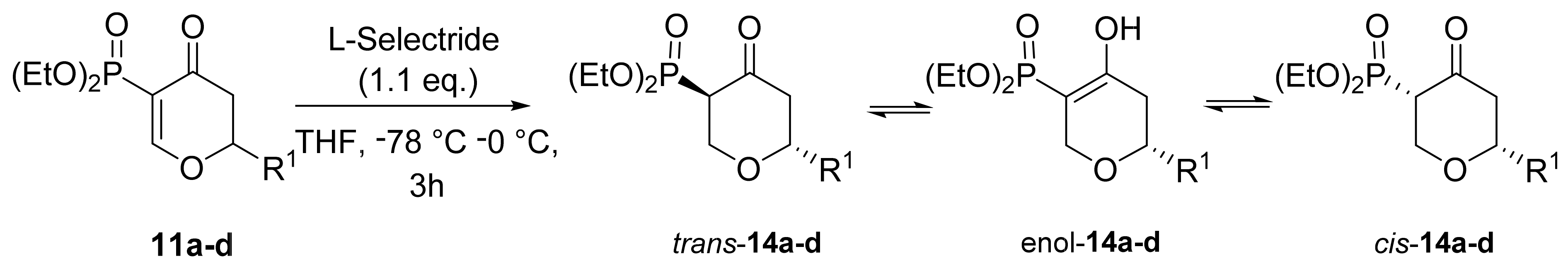
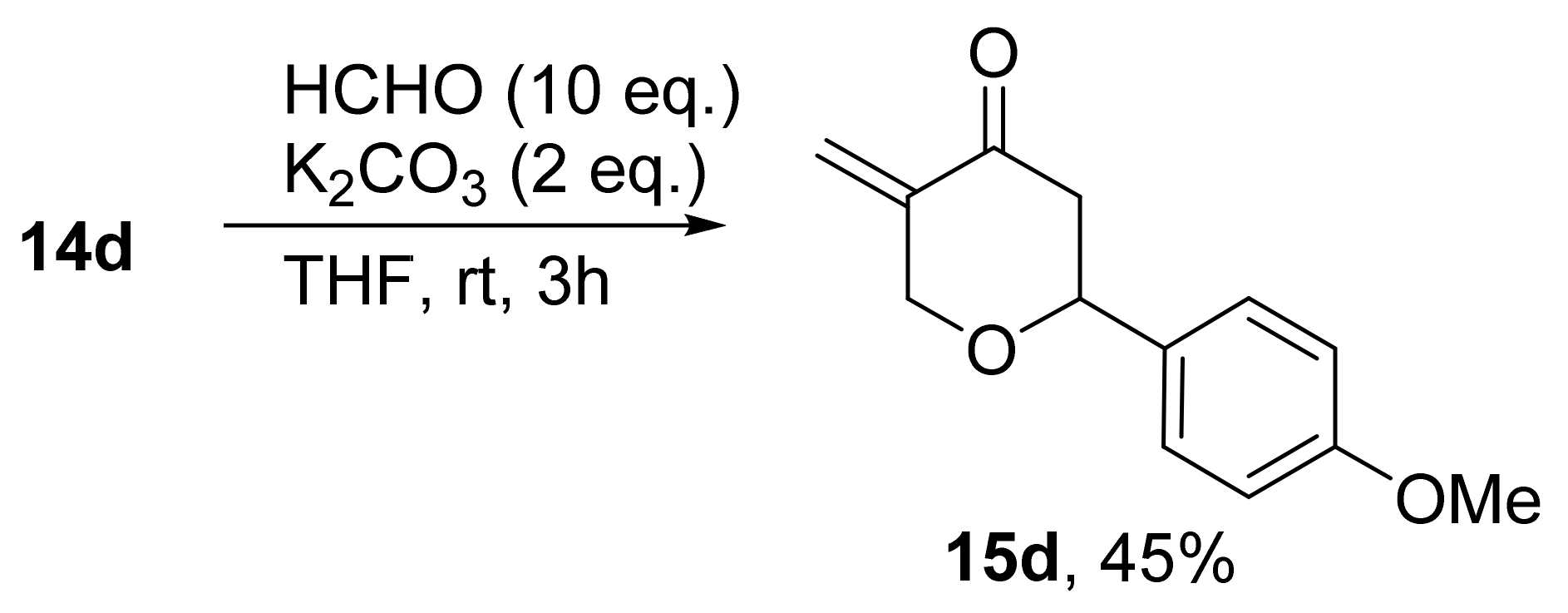
3. Results and Discussion
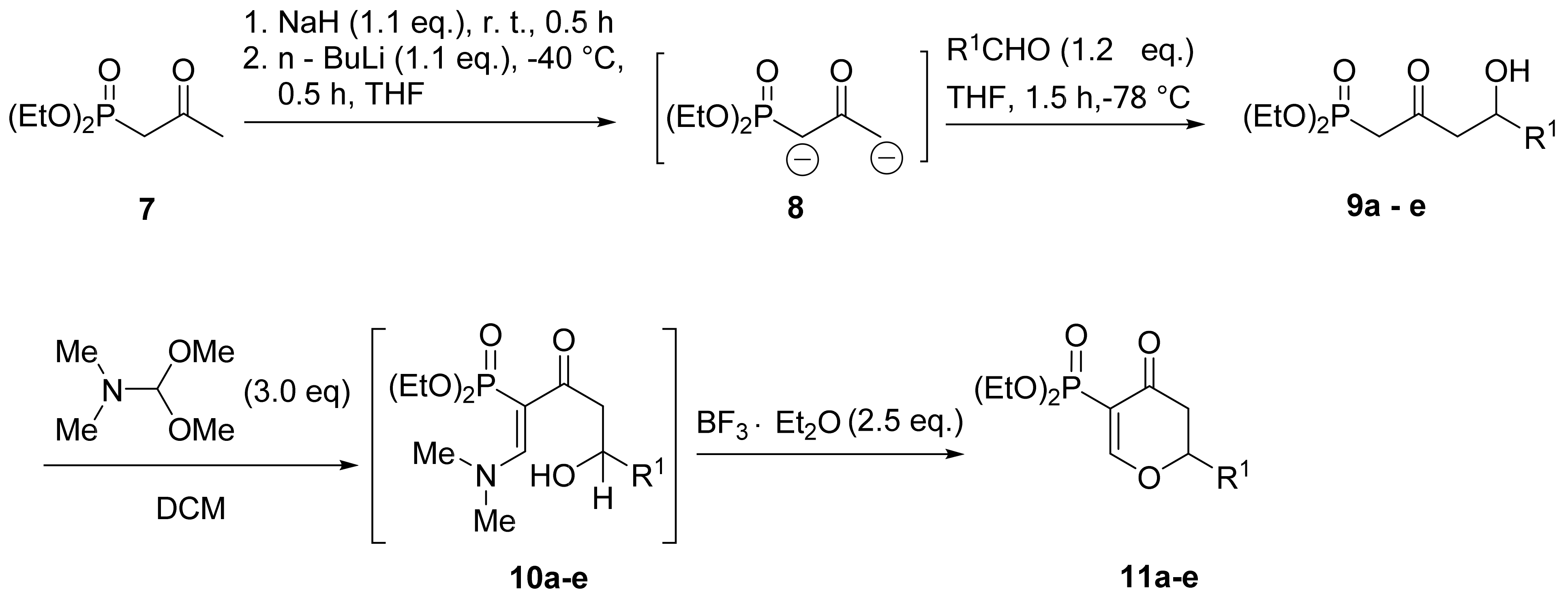
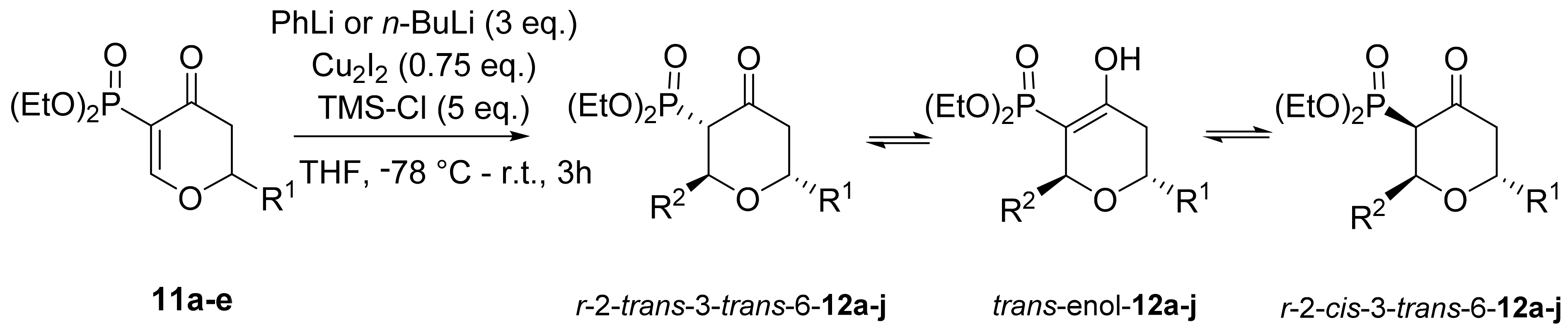
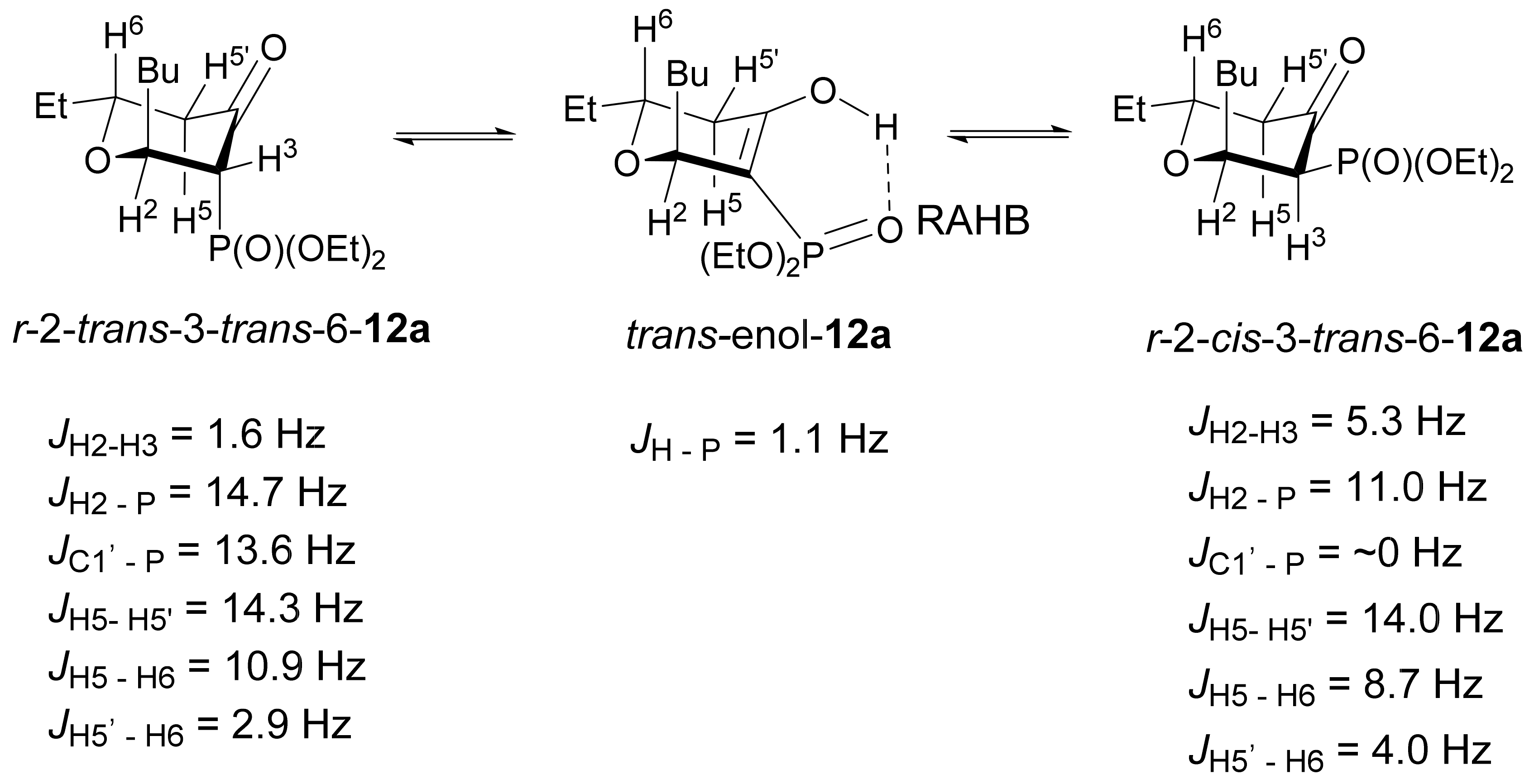
3.1. Chemistry
3.2. Biology
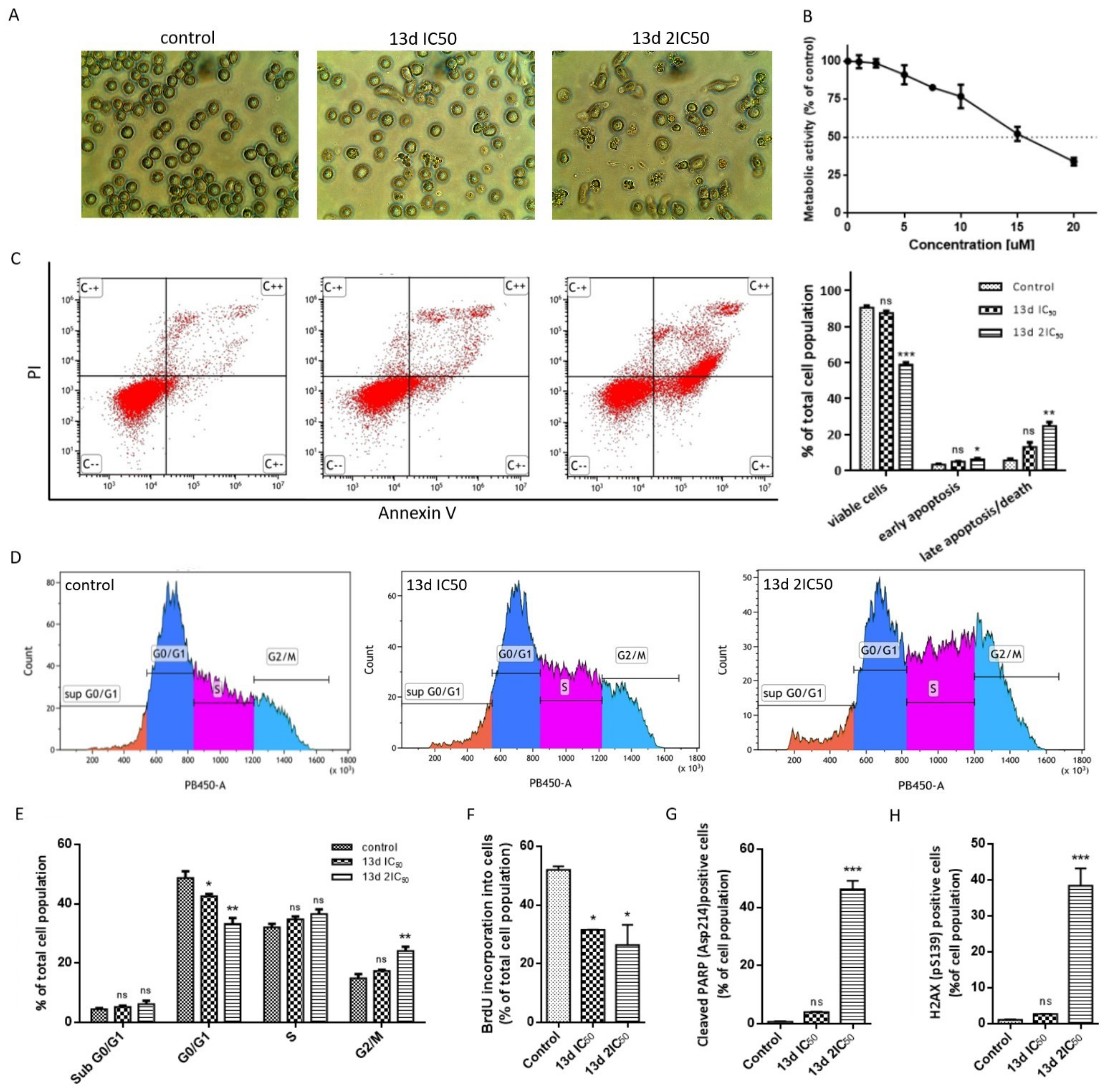
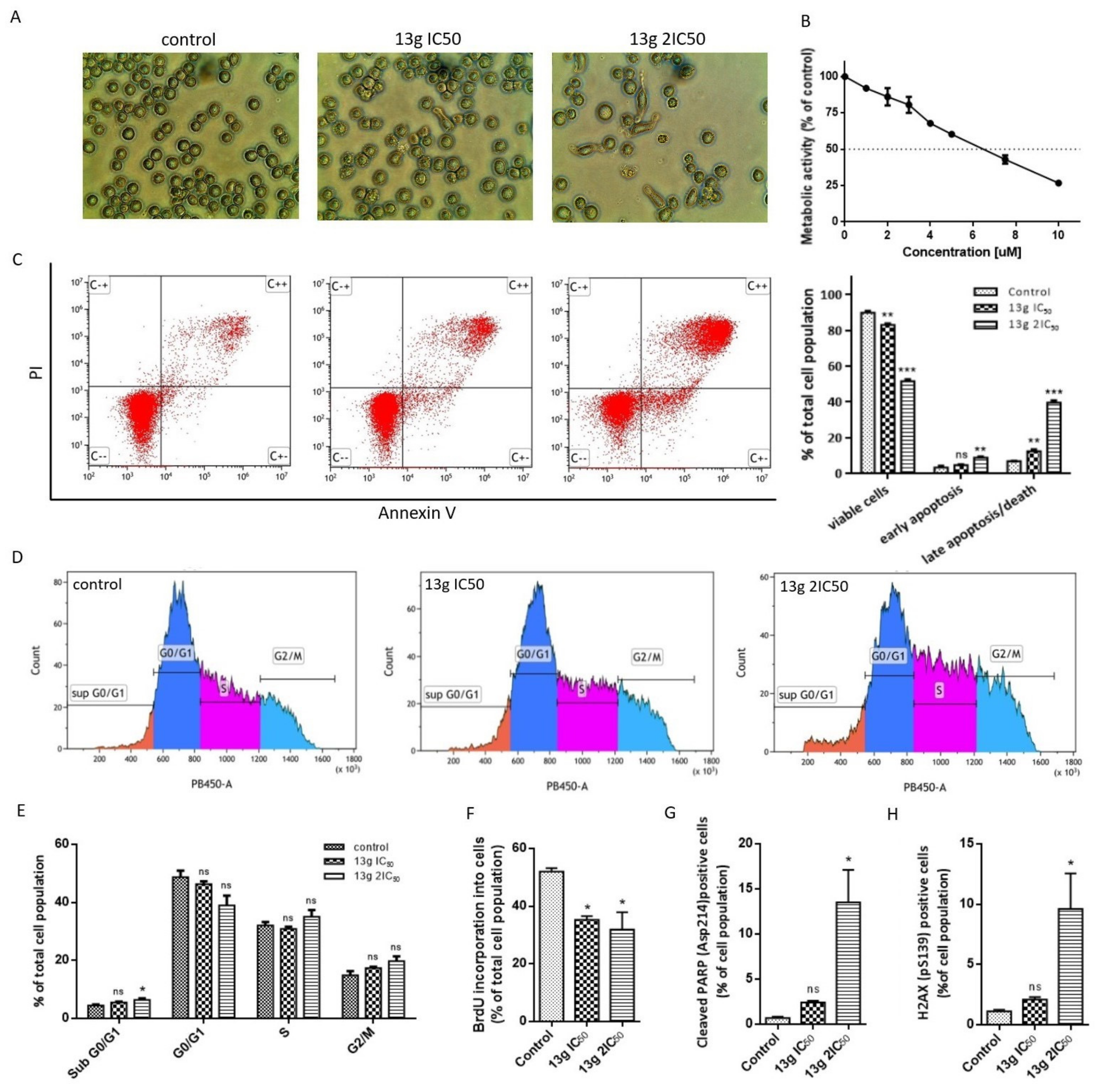
3.2.1. In Vitro Cytotoxicity
3.2.2. Antiproliferative Activity
3.2.3. Cell Morphology Changes
3.2.4. Induction of Apoptosis
3.2.5. Induction of DNA Damage
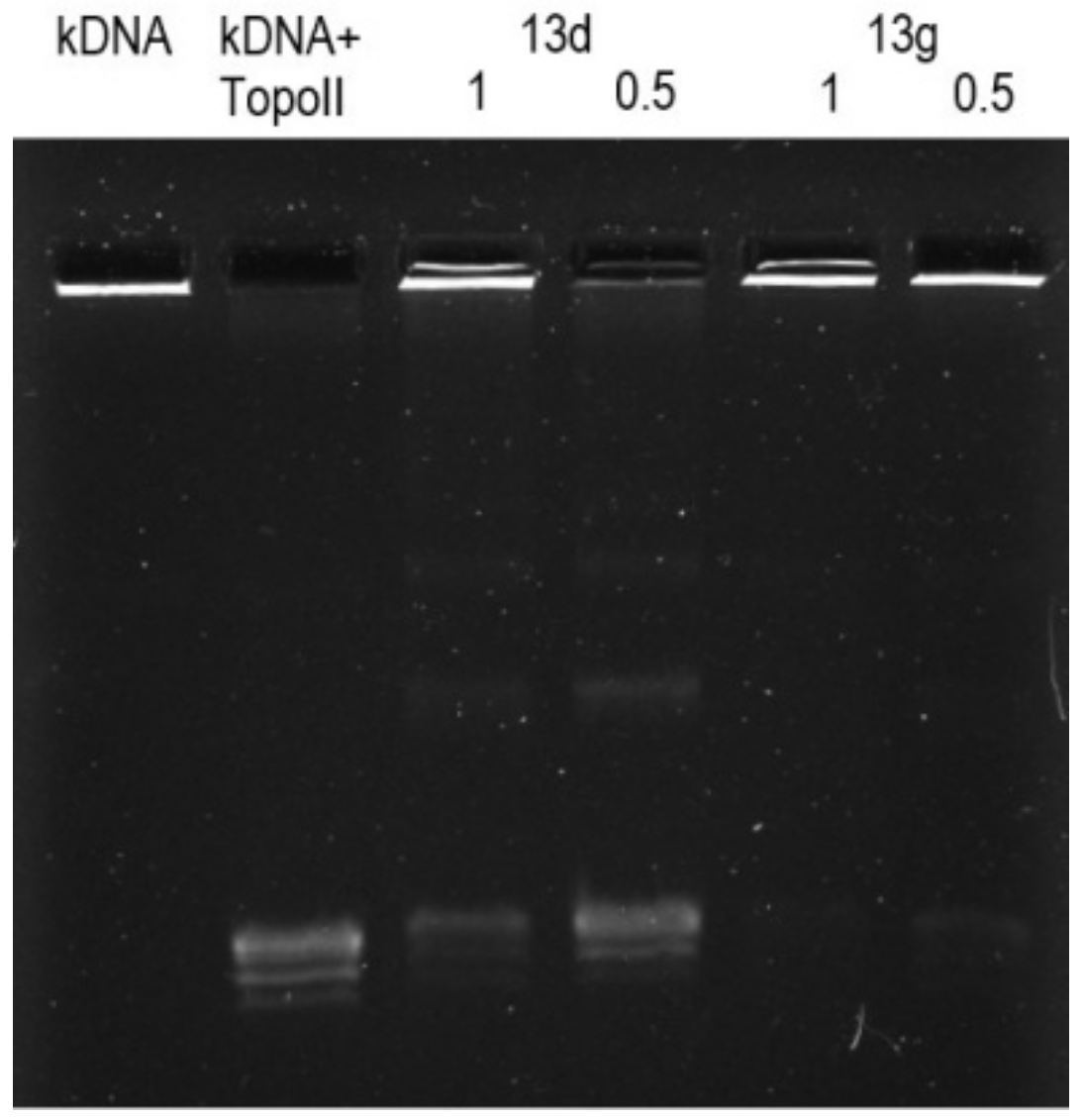
3.2.6. Topoisomerase IIα Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
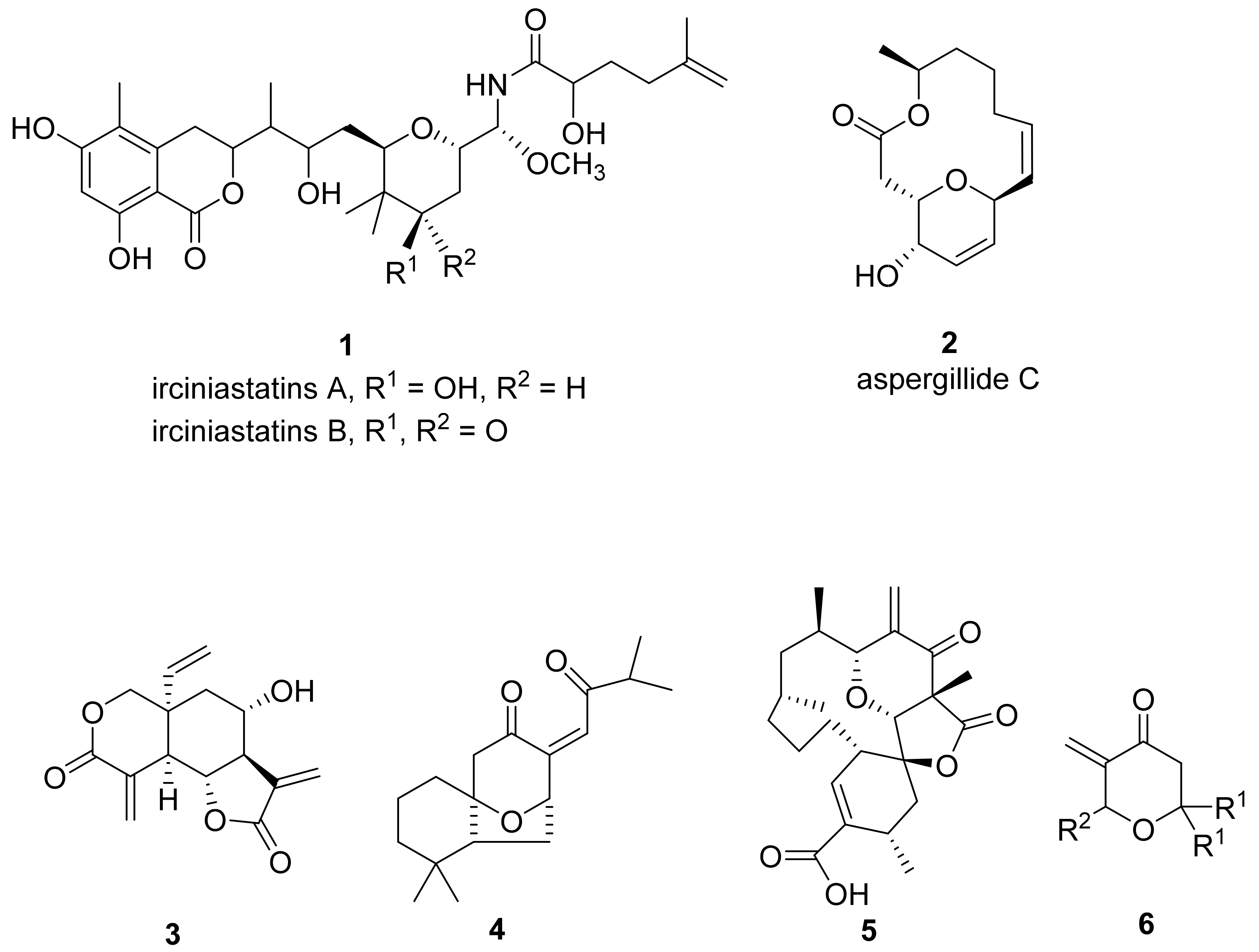
- Pettit, G.R.; Xu, J.-P.; Chapuis, J.-C.; Pettit, R.K.; Tackett, L.P.; Doubek, D.L.; Hooper, J.N.A.; Schmidt, J.M. Antineoplastic Agents. 520. Isolation and Structure of Irciniastatins A and B from the Indo-Pacific Marine Sponge Ircinia ramosa. J. Med. Chem. 2004, 47, 1149–1152. [Google Scholar] [CrossRef] [PubMed]
- Kito, K.; Ookura, R.; Yoshida, S.; Namikoshi, M.; Ooi, T.; Kusumi, T. New Cytotoxic 14-Membered Macrolides from Marine-Derived Fungus Aspergillus ostianus. Org. Lett. 2008, 10, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Janecki, T. Targets in Heterocyclic Systems; Attanasi, O.A., Spinelli, D., Eds.; Italian Society of Chemistry: Rome, 2006; Volume 10, pp. 301–320. [Google Scholar]
- Feray, L.; Bertrand, M.P. Targets in Heterocyclic Systems; Attanasi, O.A., Spinelli, D., Eds.; Italian Society of Chemistry: Rome, 2008; Volume 12, pp. 31–58. [Google Scholar]
- Kitson, R.R.A.; Millemaggi, A.; Taylor, R.J.K. The Renaissance of α-Methylene-γ-butyrolactones: New Synthetic Approaches. Angew. Chem. Int. Ed. 2009, 48, 9426–9451. [Google Scholar] [CrossRef] [PubMed]
- Elford, T.G.; Hall, D.G. Advances in 2-(Alkoxycarbonyl)allylboration of Carbonyl Compounds and Other Direct Methods for the Preparation of α-Exo-Alkylidene γ-Lactones. Synthesis 2010, 2010, 893–907. [Google Scholar] [CrossRef]
- Jiang, Z.-Y.; Zhou, J.; Huang, C.-G.; Hu, Q.-F.; Huang, X.-Z.; Wang, W.; Zhang, L.-Z.; Li, G.-P.; Xia, F.-T. Two novel antiviral terpenoids from the cultured Perovskia atriplicifolia. Tetrahedron 2015, 71, 3844–3849. [Google Scholar] [CrossRef]
- Imai, H.; Suzuki, K.-I.; Morioka, M.; Numasaki, Y.; Kadota, S.; Nagai, K.; Sato, T.; Iwanami, M.; Saito, T. Okilactomycin, a novel antibiotic produced by a Streptomyces species. J. Antibiot. 1987, 40, 1475–1482. [Google Scholar] [CrossRef] [Green Version]
- Bartosik, T.; Kędzia, J.; Drogosz-Stachowicz, J.; Janecka, A.; Krajewska, U.; Mirowski, M.; Janecki, T. Synthesis of 2,2,6-Trisubstituted 5-Methylidenetetrahydropyran-4-ones with Anticancer Activity. Molecules 2020, 25, 611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouda, M.F.R.; Abd-Elzaher, M.M.; Abdelsamaia, R.A.; Labib, A.A. On the medicinal chemistry of ferrocene. Appl. Organomet. Chem. 2007, 21, 613–625. [Google Scholar] [CrossRef]
- Ornelas, C. Application of ferrocene and its derivatives in cancer research. New J. Chem. 2011, 35, 1973–1985. [Google Scholar] [CrossRef]
- Gasser, G.; Metzler-Nolte, N. The potential of organometallic complexes in medicinal chemistry. Curr. Opin. Chem. Biol. 2012, 16, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Lal, B.; Badshah, A.; Altaf, A.A.; Khan, N.; Ullah, S. Miscellaneous applications of ferrocene-based peptides/amides. Appl. Organomet. Chem. 2011, 25, 843–855. [Google Scholar] [CrossRef]
- Hillard, E.; Vessieres, A.; Thouin, L.; Jaouen, G.; Amatore, C. Ferrocene-Mediated Proton-Coupled Electron Transfer in a Series of Ferrocifen-Type Breast-Cancer Drug Candidates. Angew. Chem. Int. Ed. 2006, 45, 285–290. [Google Scholar] [CrossRef] [PubMed]
- James, P.; Neudorfl, J.; Eissmann, M.; Jesse, P.; Prokop, A.; Schmalz, H.G. Enantioselective Synthesis of Ferrocenyl Nucleoside Analogues with Apoptosis-Inducing Activity. Org. Lett. 2006, 8, 2763–2766. [Google Scholar] [CrossRef] [PubMed]
- Jaouen, G.; Top, S.; Vessieres, A.; Leclercq, G.; Mc, J.M. The First Organometallic Selective Estrogen Receptor Modulators (SERMs) and Their Relevance to Breast Cancer. Curr. Med. Chem. 2004, 11, 2505–2517. [Google Scholar] [CrossRef] [PubMed]
- Forgues, S.F.; Nicot, B.D. Ferrocene and ferrocenyl derivatives in luminescent systems. J. Photochem. Photobiol. A Chem. 2000, 132, 137–159. [Google Scholar] [CrossRef]
- Nguyen, P.; Elipe, P.G.; Manners, I. Organometallic Polymers with Transition Metals in the Main Chain. Chem. Rev. 1999, 99, 1515–1548. [Google Scholar] [CrossRef]
- Biot, C.; Glorian, G.; Maciejewski, L.A.; Brocard, J.S.; Domarle, O.; Blampain, G.; Millet, P.; Georges, A.J.; Abessolo, H.; Dive, D.; et al. Synthesis and Antimalarial Activity in Vitro and in Vivo of a New Ferrocene-Chloroquine Analogue. J. Med. Chem. 1997, 40, 3715–3718. [Google Scholar] [CrossRef]
- Nguyen, A.; Vessieres, A.; Hillard, E.A.; Top, S.; Pigeon, P.; Jaouen, J. Ferrocifens and Ferrocifenols as New Potential Weapons against Breast Cancer. Chimia 2007, 61, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Wada, E.; Kanemasa, S.; Tsuge, O. New Synthesis of 2-Oxo-3-alkenylphosphonates and Hetero Diels–Alder Reactions with Vinyl Ethers Leading to 5-Substituted 2-Phosphinyl-2-cyclohexen-1-ones. Bull. Chem. Soc. Jpn. 1989, 62, 860–868. [Google Scholar] [CrossRef]
- Diethyl 2-oxopropylphosphonate 7 was purchased from Merck®.
- Koszuk, J.; Bartosik, T.; Wojciechowski, J.; Wolf, W.M.; Janecka, A.; Drogosz, J.; Długosz, A.; Krajewska, U.; Mirowski, M.; Janecki, T. Synthesis of 3-Methylidene-1-tosyl-2,3-dihydroquinolin-4-(1H)-ones as Potent Cytotoxic Agents. Chem. Biodivers. 2018, 15, e1800242. [Google Scholar] [CrossRef]
- Hou, T.; Raftery, D. Analysis and Purification Methods in Combinatorial Chemistry; Yan, B., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2004; Volume 163, pp. 137–173. [Google Scholar]
- Schwartz, G.K.; Shah, M.A. Targeting the cell cycle: A new approach to cancer therapy. J. Clin. Oncol. 2005, 23, 9408–9421. [Google Scholar] [CrossRef] [PubMed]
- Tyurina, Y.Y.; Shvedova, A.A.; Kawai, K.; Tyurin, V.A.; Kommineni, C.; Quinn, P.J.; Kagan, V.E. Phospholipid signaling in apoptosis: Peroxidation and externalization of phosphatidylserine. Toxicology 2000, 148, 93–101. [Google Scholar] [CrossRef]
- Van Engeland, M.; Nieland, L.J.; Ramaekers, F.C.; Schutte, B.; Reutelingsperger, C.P. Annexin V-affinity assay: A review on an apoptosis detection system based on phosphatidylserine exposure. Cytom. J. Int. Soc. Anal. Cytol. 1998, 31, 1–9. [Google Scholar] [CrossRef]
- Mashimo, M.; Onishi, M.; Uno, A.; Tanimichi, A.; Nobeyama, A.; Mori, M.; Fujii, T. The 89-kDa PARP1 cleavage fragment serves as a cytoplasmic PAR carrier to induce AIF-mediated apoptosis. J. Biol. Chem. 2021, 296, 100046. [Google Scholar] [CrossRef]
- Jackson, S.P. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002, 23, 687–696. [Google Scholar] [CrossRef]
- Aparicio, T.; Baer, R.; Gautier, J. DNA double-strand break repair pathway choice and cancer. DNA Repair 2014, 19, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Stope, M.B. Phosphorylation of histone H2A. X as a DNA-associated biomarker. World Acad. Sci. J. 2021, 3, 31. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA damage response in cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Dehshahri, A.; Ashrafizadeh, M.; Afshar, E.G.; Pardakhty, A.; Mandegary, A.; Mohammadinejad, R.; Sethi, G. Topoisomerase inhibitors: Pharmacology and emerging nanoscale delivery systems. Pharmacol. Res. 2020, 151, 104551. [Google Scholar] [CrossRef]
- Marinello, J.; Delcuratolo, M.; Capranico, G. Anthracyclines as topoisomerase II poisons: From early studies to new perspectives. Int. J. Mol. Sci. 2018, 19, 3480. [Google Scholar] [CrossRef] [Green Version]
- Larsen, A.K.; Escargeuil, A.E.; Skladanowski, A. From DNA damage to G~ 2 arrest: The many roles of topoisomerase II. Prog. Cell Cycle Res. 2003, 5, 295–300. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R1 | Yield [%] a |
|---|---|---|
| 9 | ||
| a | Et | 71 |
| b | i-Pr | 73 |
| c | Ph | 76 b |
| d | p-MeOPh | 84 b |
| e |  | 80 |
| 12 | R1 | R2 | r-2-trans-3-trans-6 a | trans-enol a | r-2-cis-3- trans-6 a | Yield b [%] |
|---|---|---|---|---|---|---|
| a | Et | n-Bu | 81 | 11 | 8 | 65 |
| b | Ph | 67 | 29 | 4 | 84 | |
| c | i-Pr | n-Bu | 77 | 12 | 11 | 76 |
| d | Ph | 72 | 27 | 1 | 97 | |
| e | Ph | n-Bu | 52 | 46 | 2 | 88 |
| f | Ph | 62 | 36 | 2 | 89 | |
| g | p-MeOPh | n-Bu | 87 | 11 | 2 | 73 |
| h | Ph | 72 | 27 | 1 | 81 | |
| i | 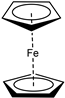 | n-Bu | 74 | 14 | 12 | 79 |
| j | Ph | 55 | 42 | 3 | 63 |
| 12R1 | R1 | r-2-trans-3-trans-6 a | trans-enol a | r-2-cis-3- trans-6 a | r-2-trans-3- cis-6 a | cis-enol a | r-2-cis-3- cis-6 a | Yield b [%] |
|---|---|---|---|---|---|---|---|---|
| k | Et | 46 | 1 | 1 | 27 | 3 | 22 | 63 |
| l | i-Pr | 56 | 1 | 1 | 29 | 2 | 11 | 68 |
| m | Ph | 57 | 5 | 1 | 23 | 1 | 13 | 87 |
| n | p-MeOPh | 44 | 2 | 1 | 40 | 3 | 10 | 67 |
| o | 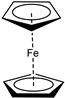 | 81 | 5 | 1 | 4 | 1 | 8 | 59 |
| 13 | R1 | R2 | Yield a [%] |
|---|---|---|---|
| a | Et | i-Pr | 44 |
| b | n-Bu | 88 | |
| c | Ph | 67 | |
| d | i-Pr | i-Pr | 50 |
| e | n-Bu | 83 | |
| f | Ph | 52 | |
| g | Ph | i-Pr | 48 |
| h | n-Bu | 77 | |
| i | Ph | 60 | |
| j | p-MeOC6H4 | i-Pr | 41 |
| k | n-Bu | 82 | |
| l | Ph | 50 | |
| m | 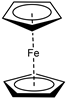 | i-Pr | 32 |
| n | n-Bu | 60 | |
| o | Ph | 52 |
| 14 | R1 | transa | enol a | cisa | Yield b [%] |
|---|---|---|---|---|---|
| a | Et | 52 | 22 | 26 | 71 |
| b | i-Pr | 55 | 11 | 34 | 76 |
| c | Ph | 52 | 13 | 35 | 74 |
| d | p-MeOC6H4 | 62 | 27 | 11 | 62 |
| Analog | R1 | R2 | IC50 [μM] a | ||
|---|---|---|---|---|---|
| HL-60 | MCF-7 | HUVEC | |||
| Et | |||||
| 13a | i-Pr | 21.30 ± 1.00 | 20.36 ± 0.35 | ||
| 13b | n-Bu | 39.15 ± 0.15 | 106.5 ± 0.50 | ||
| 13c | Ph | 11.85 ± 0.15 | 36.00 ± 0.41 | ||
| i-Pr | |||||
| 13d | i-Pr | 7.65 ± 0.05 | 14.00 ± 1.00 | 24.7 ± 0.65 | |
| 13e | n-Bu | 78.75 ± 5.25 | >150 | ||
| 13f | Ph | 8.45 ± 0.05 | 69.65 ± 2.16 | 11.94 ± 2.32 | |
| Ph | |||||
| 13g | i-Pr | 3.69 ± 0.08 | 8.63 ± 0.07 | 3.84 ± 0.01 | |
| 13h | n-Bu | 134.00 ± 1.00 | >150 | ||
| 13i | Ph | 36.00 ± 1.41 | >200 | ||
| p-MeOC6H4 | |||||
| 13j | i-Pr | 3.75 ± 0.07 | 9.11 ± 0.02 | 4.25 ± 0.05 | |
| 13k | n-Bu | >100 | >100 | ||
| 13l | Ph | 25.01 ± 0.94 | 203.00 ± 56.00 | ||
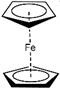 | |||||
| 13m | i-Pr | 3.87 ± 0.16 | 5.73 ± 0.36 | ||
| 13n | n-Bu | 62.60 ± 0.61 | >100 | ||
| 13o | Ph | >100 | >100 | ||
| 15d | p-MeOC6H4 | H | 163.00 ± 7.07 | >200 | |
| Carboplatin | 2.53 ± 0.20 | 3.76 ± 0.35 | 5.35 ± 0.05 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartosik, T.; Drogosz-Stachowicz, J.; Janecka, A.; Kędzia, J.; Pacholczyk-Sienicka, B.; Szymański, J.; Gach-Janczak, K.; Janecki, T. Stereoselective Synthesis and Anticancer Activity of 2,6-Disubstituted trans-3-Methylidenetetrahydropyran-4-ones. Materials 2022, 15, 3030. https://doi.org/10.3390/ma15093030
Bartosik T, Drogosz-Stachowicz J, Janecka A, Kędzia J, Pacholczyk-Sienicka B, Szymański J, Gach-Janczak K, Janecki T. Stereoselective Synthesis and Anticancer Activity of 2,6-Disubstituted trans-3-Methylidenetetrahydropyran-4-ones. Materials. 2022; 15(9):3030. https://doi.org/10.3390/ma15093030
Chicago/Turabian StyleBartosik, Tomasz, Joanna Drogosz-Stachowicz, Anna Janecka, Jacek Kędzia, Barbara Pacholczyk-Sienicka, Jacek Szymański, Katarzyna Gach-Janczak, and Tomasz Janecki. 2022. "Stereoselective Synthesis and Anticancer Activity of 2,6-Disubstituted trans-3-Methylidenetetrahydropyran-4-ones" Materials 15, no. 9: 3030. https://doi.org/10.3390/ma15093030
APA StyleBartosik, T., Drogosz-Stachowicz, J., Janecka, A., Kędzia, J., Pacholczyk-Sienicka, B., Szymański, J., Gach-Janczak, K., & Janecki, T. (2022). Stereoselective Synthesis and Anticancer Activity of 2,6-Disubstituted trans-3-Methylidenetetrahydropyran-4-ones. Materials, 15(9), 3030. https://doi.org/10.3390/ma15093030