
A New BCN Compound with Monoclinic Symmetry: First-Principle Calculations

Abstract
:1. Introduction
2. Theoretical Methods
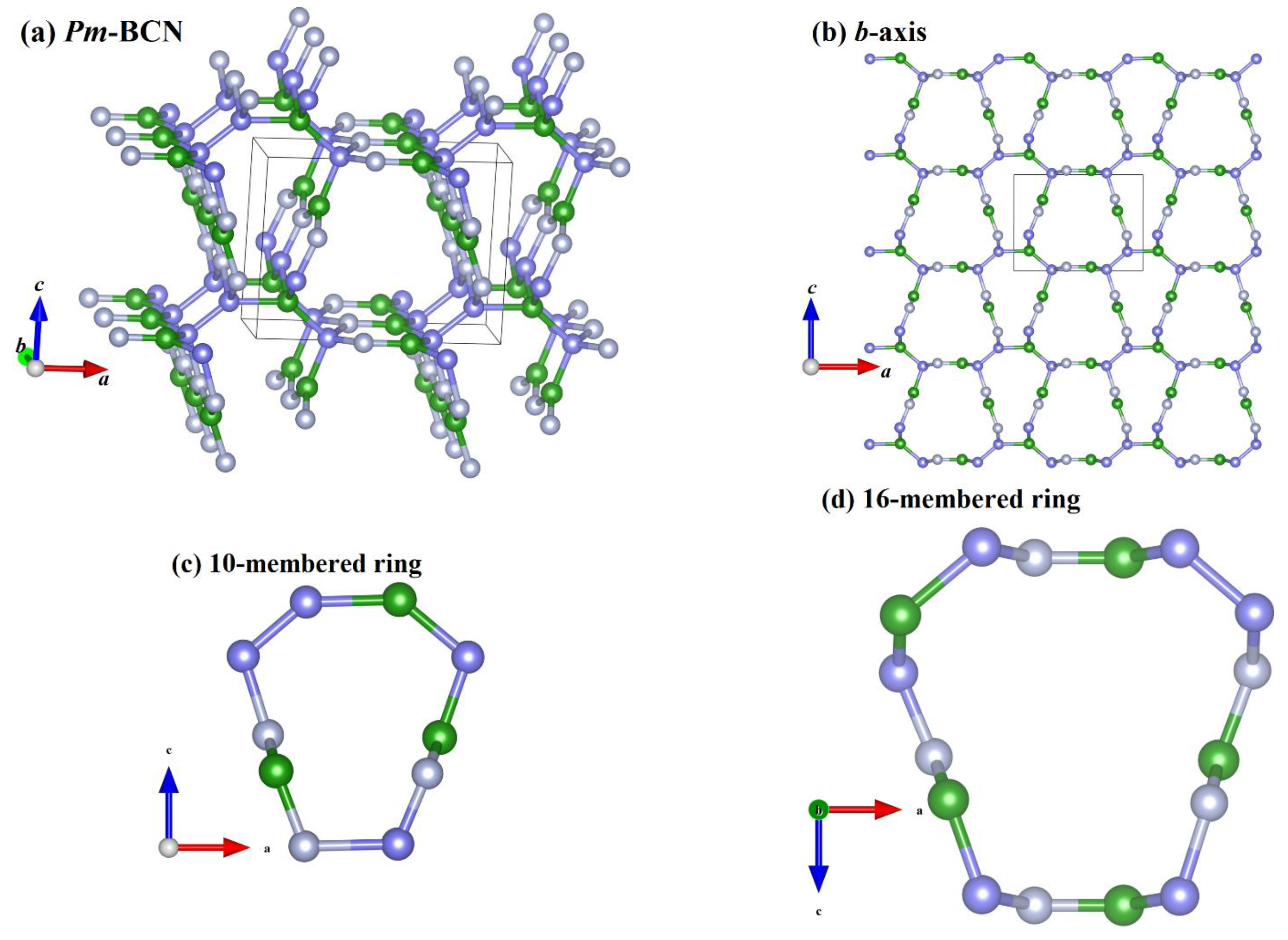
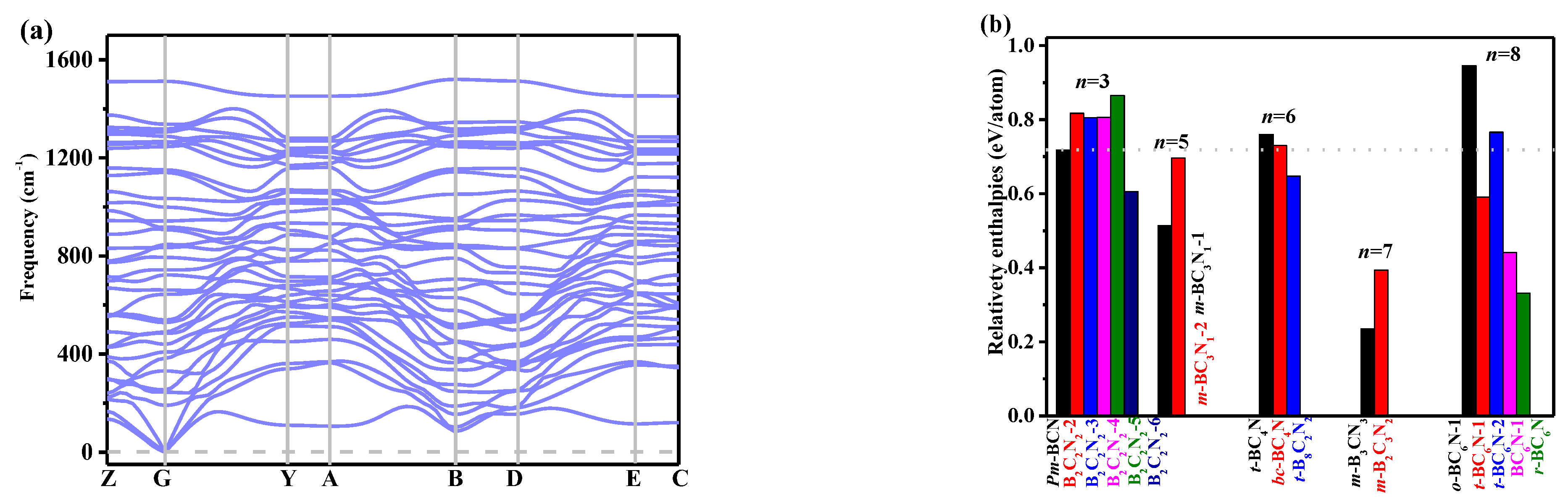
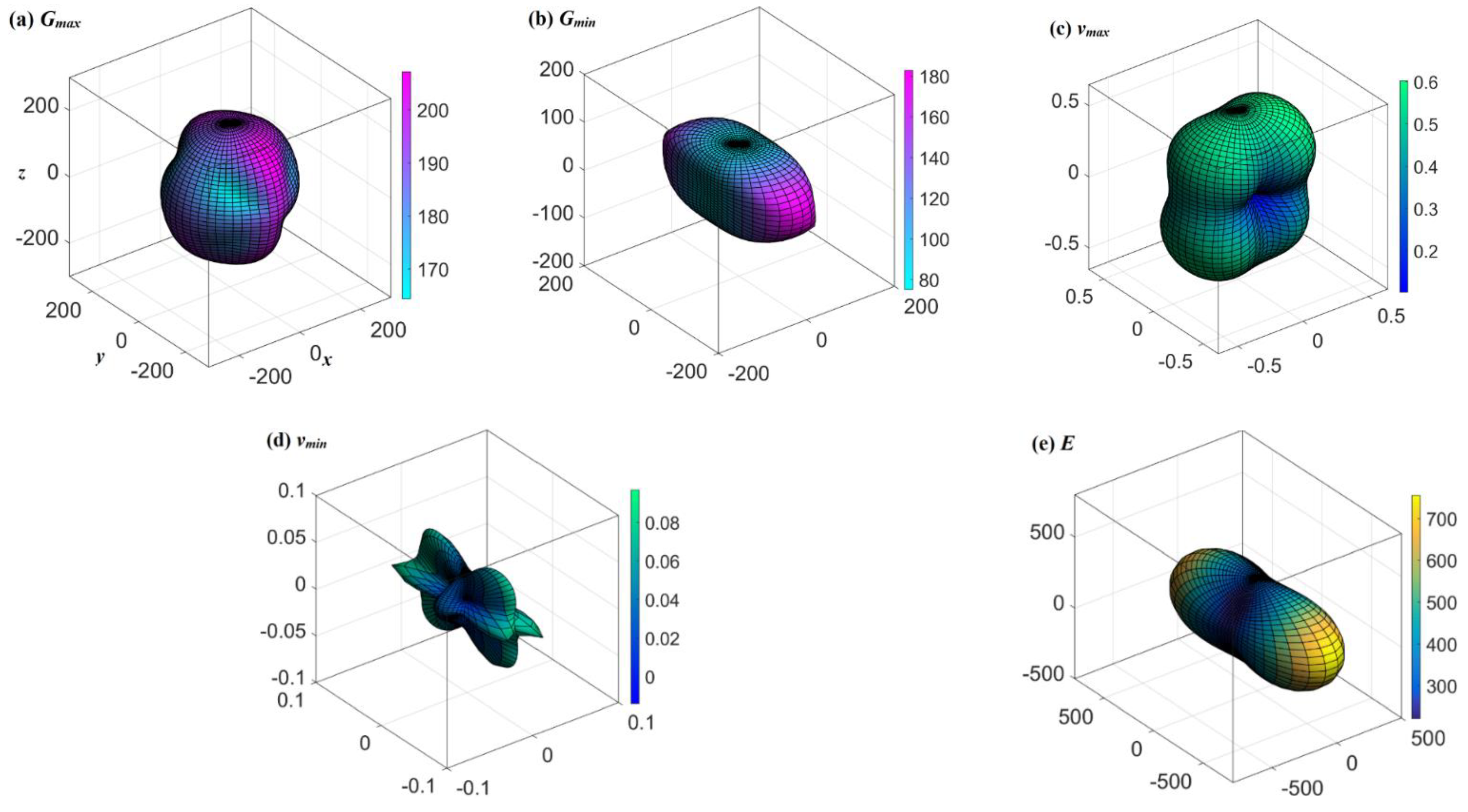
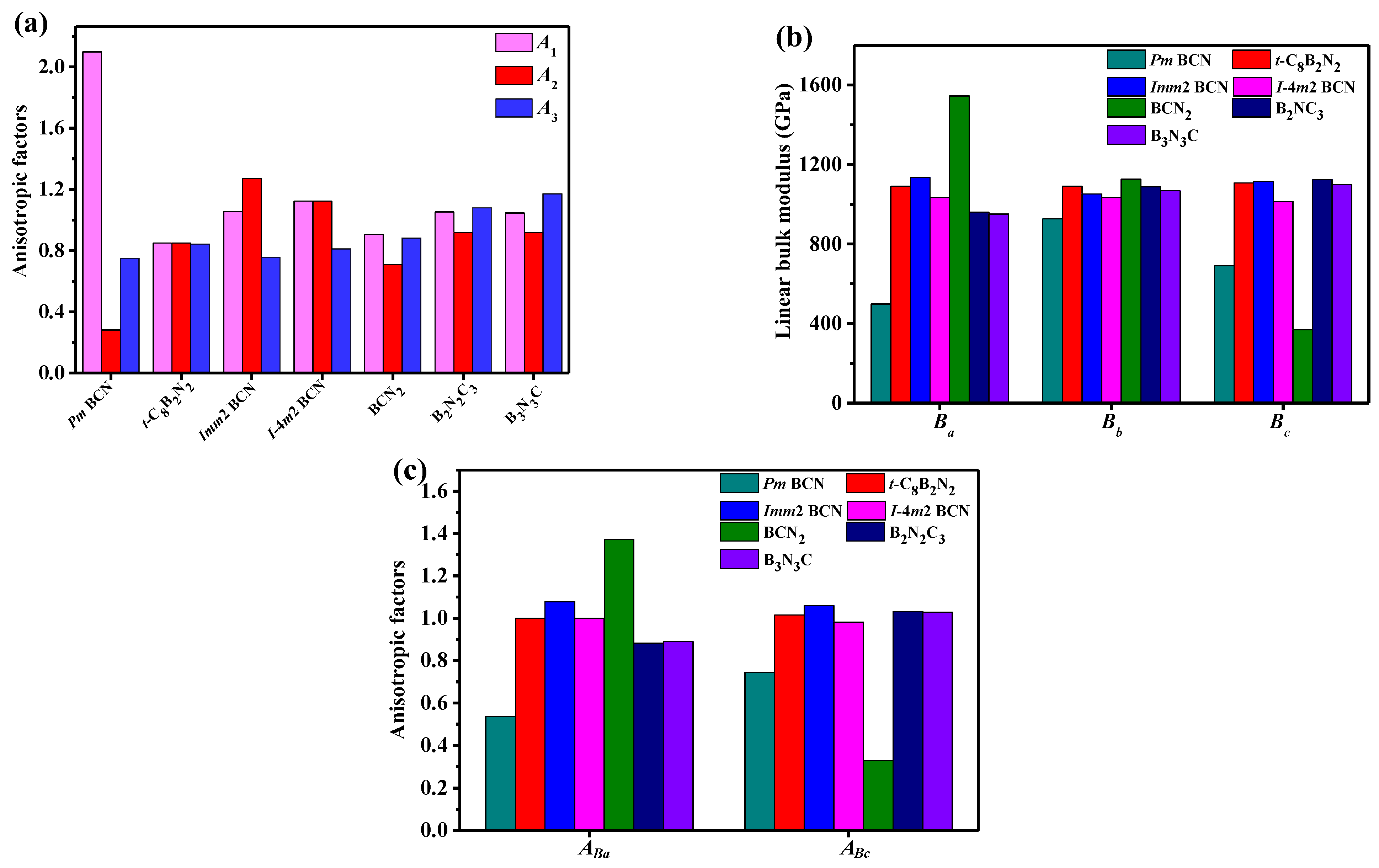
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fan, Q.; Liu, H.; Jiang, L.; Zhang, W.; Song, Y.; Wei, Q.; Yu, X.; Yun, S. Three-dimensional metallic carbon allotropes with superhardness. Nanotechnol. Rev. 2021, 10, 1266–1276. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, Y.; Huang, Q.; Ma, M.; Pan, Y.; Xiong, M.; Li, Z.; Zhao, Z.; He, J.; Yu, D. First principles studies of superhard BC6N phases with unexpected 1D metallicity. Comput. Mater. Sci. 2018, 148, 157–164. [Google Scholar] [CrossRef]
- Fan, Q.; Wu, N.; Yang, R.; Zhang, W.; Yu, X.; Yun, S. All sp2 hybridization BN polymorphs with wide bandgap. J. Appl. Phys. 2022, 131, 055703. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; Xing, M.; Jin, J. Novel BCN2 and CN compounds in C2/m phase: First-principle calculations. J. Phys. Chem. Solids 2021, 158, 110231. [Google Scholar] [CrossRef]
- Li, X.; Xing, M. Two novel superhard monoclinic phase of B–C–N compounds. J. Solid State Chem. 2020, 292, 121750. [Google Scholar] [CrossRef]
- Fan, Q.; Li, C.; Yang, R.; Yu, X.; Zhang, W.; Yun, S. Stability, mechanical, anisotropic and electronic properties of oP8 carbon: A superhard carbon allotrope in orthorhombic phase. J. Solid State Chem. 2021, 294, 121894. [Google Scholar] [CrossRef]
- Qu, N.R.; Wang, H.C.; Li, Q.; Li, Y.D.; Li, Z.P.; Gou, H.Y.; Gao, F.M. Surperhard monoclinic BC6N allotropes: First-principles investigations. Chin. Phys. B 2019, 28, 096201. [Google Scholar] [CrossRef]
- Li, S.; Shi, L. Two novel superhard structures: Monoclinic BC3N. Physica B 2020, 584, 412061. [Google Scholar] [CrossRef]
- Fan, Q.; Liu, H.; Jiang, L.; Yu, X.; Zhang, W.; Yun, S. Two orthorhombic superhard carbon allotropes: C16 and C24. Diam. Relat. Mater. 2021, 116, 108426. [Google Scholar] [CrossRef]
- Zhu, H.; Shi, L.; Li, S.; Duan, Y.; Zhang, S.; Xia, W. Effects of hydrostatic pressure and biaxial strains on the elastic and electronic properties of t-C8B2N2. J. Appl. Phys. 2018, 123, 135103. [Google Scholar] [CrossRef]
- Wang, D.; Shi, R.; Gan, L.H. t-C8B2N2: A potential superhard material. Chem. Phys. Lett. 2017, 669, 80–84. [Google Scholar] [CrossRef]
- Fan, Q.; Liu, H.; Yang, R.; Yu, X.; Zhang, W.; Yun, S. An orthorhombic superhard carbon allotrope: Pmma C24. J. Solid State Chem. 2021, 300, 122260. [Google Scholar] [CrossRef]
- Wang, S.; Oganov, A.R.; Qian, G.; Zhu, Q.; Dong, H.; Dong, X.; Esfahani, M.M.D. Novel superhard B–C–O phases predicted from first principles. Phys. Chem. Chem. Phys. 2016, 18, 1859–1863. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Wei, Q.; Chai, C.; Zhang, M.; Yan, H.; Zhang, Z.; Zhang, J.; Zhang, D. Elastic and electronic properties of Imm2- and I-4m2-BCN. Comput. Mater. Sci. 2015, 97, 6–13. [Google Scholar] [CrossRef]
- Fan, Q.; Wei, Q.; Chai, C.; Yan, H.; Zhang, M.; Lin, Z.; Zhang, Z.; Zhang, J.; Zhang, D. Structural, mechanical, and electronic properties of P3m1-BCN. J. Phys. Chem. Solids 2015, 79, 89–96. [Google Scholar] [CrossRef]
- Li, X.; Xing, M. Novel carbon-rich nitride C3N: A superhard phase in monoclinic symmetry. Comput. Mater. Sci. 2019, 158, 170–177. [Google Scholar] [CrossRef]
- Xing, M.; Li, X. BC2O in C2/m phase: Light element compound with direct band gaps. J. Solid State Chem. 2021, 304, 122590. [Google Scholar] [CrossRef]
- Fan, Q.; Wei, Q.; Chai, C.; Yu, X.; Liu, Y.; Zhou, P.; Yan, H.; Zhang, D. First-principles Study of Structural, Elastic, Anisotropic, and Thermodynamic Properties of R3-B2C. Chin. J. Phys. 2015, 53, 100601. [Google Scholar]
- Fan, Q.; Zhao, Y.; Yu, X.; Song, Y.; Zhang, W.; Yun, S. Physical properties of a novel microporous carbon material. Diam. Relat. Mater. 2020, 106, 107831. [Google Scholar] [CrossRef]
- Cui, H.J.; Yan, Q.B.; Sheng, X.L.; Wang, D.L.; Zheng, Q.R.; Su, G. The geometric and electronic transitions in body-centered-tetragonal C8: A first principle study. Carbon 2017, 120, 89–94. [Google Scholar] [CrossRef]
- Xing, M.; Li, X. A porous nanotube network structure of metallic carbon. Results Phys. 2021, 28, 104579. [Google Scholar] [CrossRef]
- Fan, Q.; Wei, Q.; Chai, C.; Yan, H.; Zhang, M.; Zhang, Z.; Zhang, J.; Zhang, D. Structural, anisotropic and thermodynamic properties of boron carbide: First principles calculations. Indian J. Pure Ap. Phys. 2016, 54, 227–235. [Google Scholar]
- Yu, X.; Su, R.; He, B. A novel BN Polymorph with ductile manner. J. Solid State Chem. 2022, 306, 122794. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1956, 140, A1133. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Su, R.; He, B.; Ma, B. Theoretical investigations of a BN polymorph with sp2+sp3 hybridizations. Crystals 2021, 11, 1574. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Ceperley, D.M.; Alder, B.J. Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 1980, 45, 566–569. [Google Scholar] [CrossRef] [Green Version]
- Pfrommer, B.G.; Cote, M.; Louie, S.G.; Cohen, M.L. Relaxation of crystals with the quasi-Newton method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef] [PubMed]
- Baroni, S.; de Gironcoli, S.; Corso, A.D.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef] [Green Version]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef] [PubMed]
- Voigt, W. Lehrburch der Kristallphysik; Teubner, B.G., Ed.; Johnson Reprint Corp: Leipzig, Germany, 1928. (In German) [Google Scholar]
- Reuss, A. Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizit€atsbedingung für Einkristalle. Z. Angew. Math. Mech. 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. Lond. 1952, 65, 349. [Google Scholar] [CrossRef]
- Zhang, X.X.; Wang, Y.C.; Lv, J.; Zhu, C.Y.; Li, Q.; Zhang, M.; Li, Q.; Ma, Y.M. First-principles structural design of superhard materials. J. Chem. Phys. 2013, 138, 114101. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Li, L.; Wang, W.; Tian, Y. Superhard B2C2N2 compounds from first-principles calculations. J. Appl. Phys. 2011, 109, 023516. [Google Scholar]
- Wu, Z.J.; Zhao, E.J.; Xiang, H.P.; Hao, X.F.; Liu, X.J.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Wu, N.; Chen, S.M.; Jiang, L.; Zhang, W.; Yu, X.H.; Yun, S.N. P213 BN: A novel large-cell boron nitride polymorph. Commun. Theor. Phys. 2021, 73, 125701. [Google Scholar] [CrossRef]
- Qiao, L.; Hang, L.; Li, P.; Zhang, H.; Yan, G. Novel Carbon-Rich B−C Compounds in orthorhombic phase: First-principles calculations. J. Solid State Chem. 2021, 300, 122263. [Google Scholar] [CrossRef]
- Dai, J.; Wu, X.; Yang, J.; Zeng, X.C. Porous Boron Nitride with Tunable Pore Size. J. Phys. Chem. Lett. 2014, 5, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Marmier, A.; Lethbridge, Z.A.D.; Walton, R.I.; Smith, C.W.; Parker, S.C.; Evans, K.E. ElAM: A computer program for the analysis and representation of anisotropic elastic properties. Comput. Phys. Commun. 2010, 181, 2102–2115. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Peng, H.C.; Zhang, W.; Yu, X.H.; Yun, S.N. Physical properties of group 14 elements in P2/m phase. J. Solid State Chem. 2022, 305, 122641. [Google Scholar] [CrossRef]
- Ma, Z.Y.; Zuo, J.; Tang, C.Z.; Wang, P.; Shi, C.L. Physical properties of a novel phase of boron nitride and its potential applications. Mater. Chem. Phys. 2020, 252, 123245. [Google Scholar] [CrossRef]
- Qiao, L.P.; Jin, Z.; Yan, G.Y.; Li, P.; Hang, L.M.; Li, L. Density-functional-studying of oP8–, tI16–, and tP4–B2CO physical properties under pressure. J. Solid State Chem. 2019, 270, 642–650. [Google Scholar] [CrossRef]
- Fan, Q.; Zhao, R.; Zhang, W.; Song, Y.; Sun, M.; Schwingenschlögl, U. Low-energy Ga2O3 polymorphs with low electron effective masses. Phys. Chem. Chem. Phys. 2022, 24, 7045–7049. [Google Scholar] [CrossRef]
- Xing, M.; Li, X. Designing sp3 networks of two novel carbon allotropes in the P4/mmm phase. Chin. J. Phys. 2022, 75, 90–103. [Google Scholar] [CrossRef]
- Fan, Q.; Hao, B.; Zhao, Y.; Song, Y.; Zhang, W.; Yun, S. Si–C alloys with direct band gaps for photoelectric application. Vacuum 2022, 199, 110952. [Google Scholar] [CrossRef]
- Xing, M.; Qian, C.; Li, X. Two novel carbon allotropes with tetragonal symmetry: First-principles calculations. J. Solid State Chem. 2022, 309, 122971. [Google Scholar] [CrossRef]
- Connétable, D.; Thomas, O. First-principles study of the structural, electronic, vibrational, and elastic properties of orthorhombic NiSi. Phys. Rev. B 2009, 79, 094101. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.Y.; Wei, Q.; Yan, H.Y.; Zhang, M.G.; Zhang, Z.X.; Zhang, J.Q.; Zhang, D.Y. Elastic and electronic properties of Pbca-BN: First-principles calculations. Comput. Mater. Sci. 2014, 85, 80–87. [Google Scholar] [CrossRef]
- Ravindran, P.; Fast, L.; Korzhavyi, P.A.; Johansson, B.; Wills, J.; Eriksson, O. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2. J. Appl. Phys. 1998, 84, 4891–4904. [Google Scholar] [CrossRef]
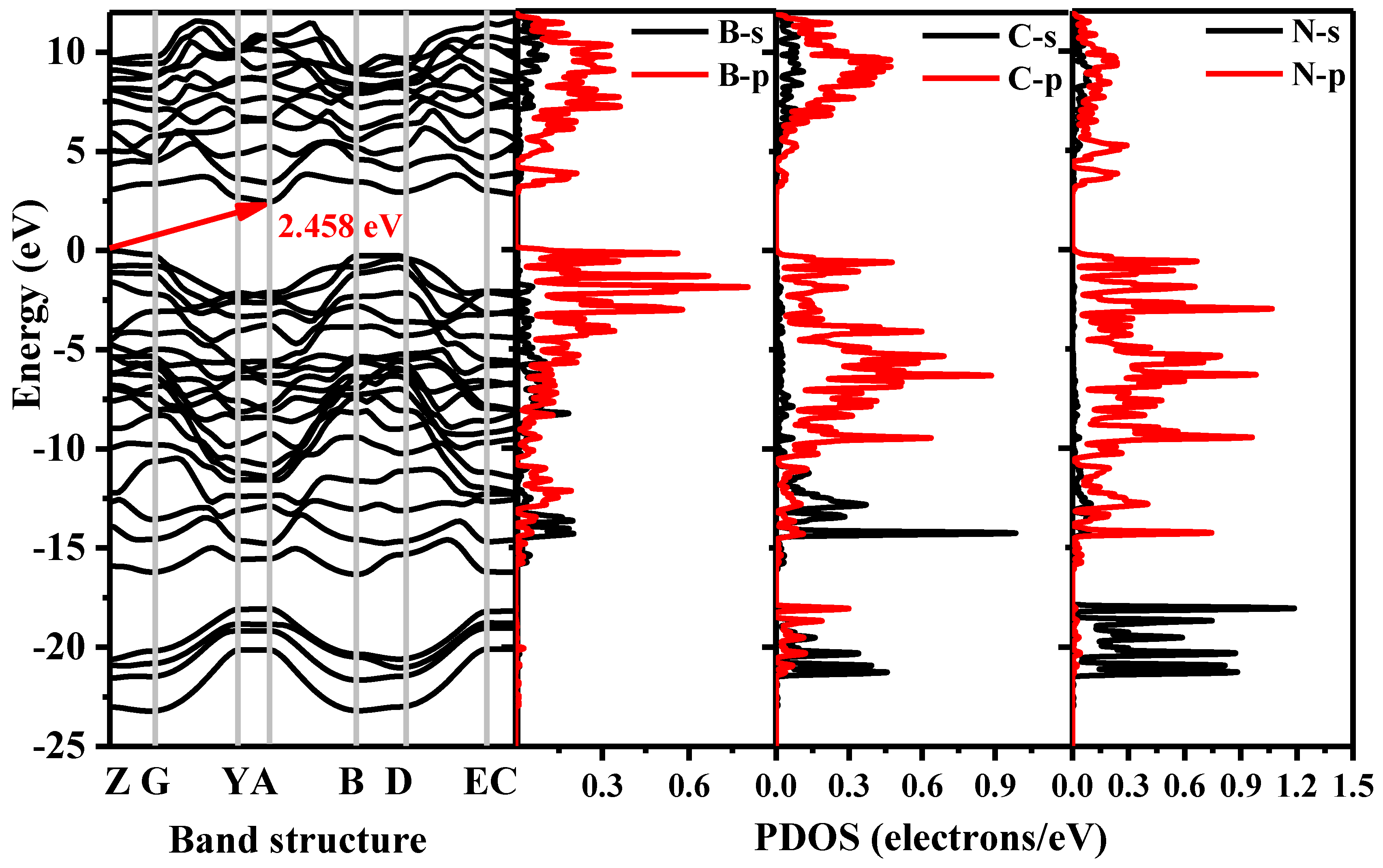
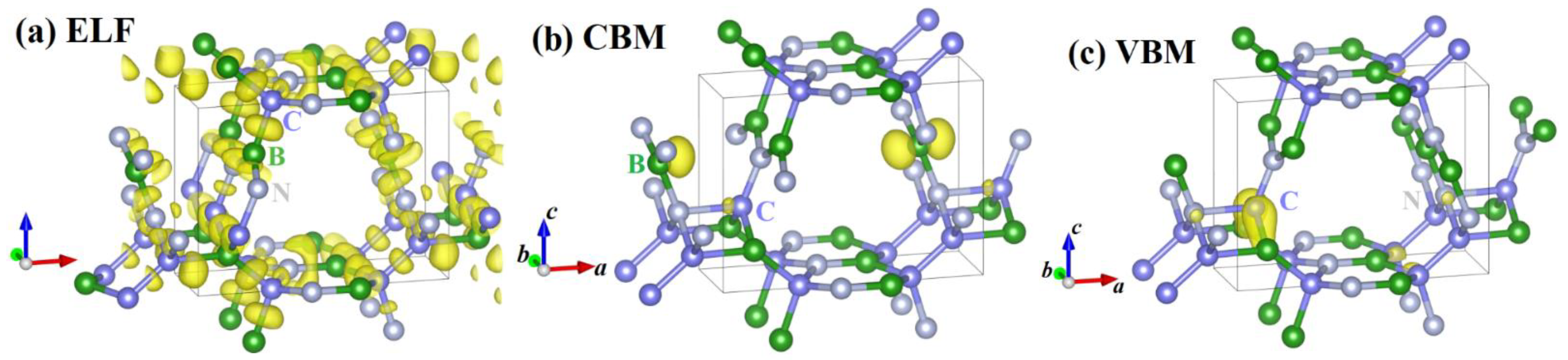

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a | b | c | β | V | ρ | ||
|---|---|---|---|---|---|---|---|
| Pm BCN | GGA | 7.2538 | 2.5387 | 5.4260 | 89.960 | 24.981 | 2.448 |
| LDA | 7.1697 | 2.5043 | 5.3334 | 89.219 | 23.938 | 2.555 | |
| t-C8B2N2 | GGA a | 2.5470 | 10.9470 | 17.781 | 3.402 | ||
| LDA b | 2.5250 | 10.8540 | 17.092 | 3.539 | |||
| Imm2 BCN | GGA | 2.5451 | 2.5658 | 10.9077 | 17.808 | 3.434 | |
| GGA c | 2.5453 | 2.5658 | 10.9169 | 17.822 | |||
| GGA d | 2.5480 | 2.5690 | 10.9130 | 17.859 | |||
| LDA | 2.5129 | 2.5313 | 10.7687 | 17.125 | 3.571 | ||
| LDA c | 2.5127 | 2.5309 | 10.7659 | 17.212 | |||
| I-4m2 BCN | GGA | 2.5648 | 10.9948 | 18.081 | 3.382 | ||
| GGA c | 2.5641 | 10.9892 | 18.063 | ||||
| GGA d | 2.5670 | 11.0020 | 18.124 | ||||
| LDA | 2.5301 | 10.8420 | 17.101 | 3.525 | |||
| LDA c | 2.5298 | 10.8396 | 17.343 |
| C11 | C12 | C13 | C15 | C22 | C23 | C25 | C33 | C35 | C44 | C46 | C55 | C66 | B | G | E | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pm BCN | GGA | 339 | 50 | 184 | 8 | 770 | 48 | 0.4 | 400 | 10 | 194 | 8 | 75 | 189 | 225 | 153 | 374 |
| LDA | 364 | 61 | 212 | 16 | 829 | 59 | 2 | 405 | 20 | 203 | 12 | 73 | 199 | 245 | 154 | 382 | |
| m-B2C3N2 | GGA a | 723 | 97 | 178 | 11 | 936 | 43 | 7 | 871 | 21 | 326 | −2 | 394 | 395 | 351 | 367 | 816 |
| m-B3CN3 | GGA a | 684 | 123 | 181 | 21 | 841 | 47 | −7 | 841 | 63 | 304 | 2 | 375 | 387 | 345 | 346 | 778 |
| t-C8 B2N2 | GGA b | 987 | 39 | 143 | 890 | 368 | 389 | 390 | 379 | 858 | |||||||
| Imm2 BCN | GGA | 963 | 23 | 144 | 898 | 141 | 395 | 395 | 457 | 343 | 367 | 394 | |||||
| GGA c | 962 | 22 | 143 | 894 | 140 | 819 | 400 | 456 | 343 | 365 | 394 | 839 | |||||
| I-4m2 BCN | GGA | 853 | 45 | 133 | 753 | 377 | 327 | 342 | 358 | ||||||||
| GGA c | 857 | 47 | 135 | 755 | 377 | 328 | 345 | 358 | 798 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Z.; Tang, C.; Shi, C. A New BCN Compound with Monoclinic Symmetry: First-Principle Calculations. Materials 2022, 15, 3186. https://doi.org/10.3390/ma15093186
Ma Z, Tang C, Shi C. A New BCN Compound with Monoclinic Symmetry: First-Principle Calculations. Materials. 2022; 15(9):3186. https://doi.org/10.3390/ma15093186
Chicago/Turabian StyleMa, Zhenyang, Chunzhi Tang, and Chunlei Shi. 2022. "A New BCN Compound with Monoclinic Symmetry: First-Principle Calculations" Materials 15, no. 9: 3186. https://doi.org/10.3390/ma15093186
APA StyleMa, Z., Tang, C., & Shi, C. (2022). A New BCN Compound with Monoclinic Symmetry: First-Principle Calculations. Materials, 15(9), 3186. https://doi.org/10.3390/ma15093186