

Preparation and Electrochromic Properties of Benzodithiophene-Isoindigo Conjugated Polymers with Oligoethylene Glycol Side Chains

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization Methods
3. Design and Synthesis
3.1. Design
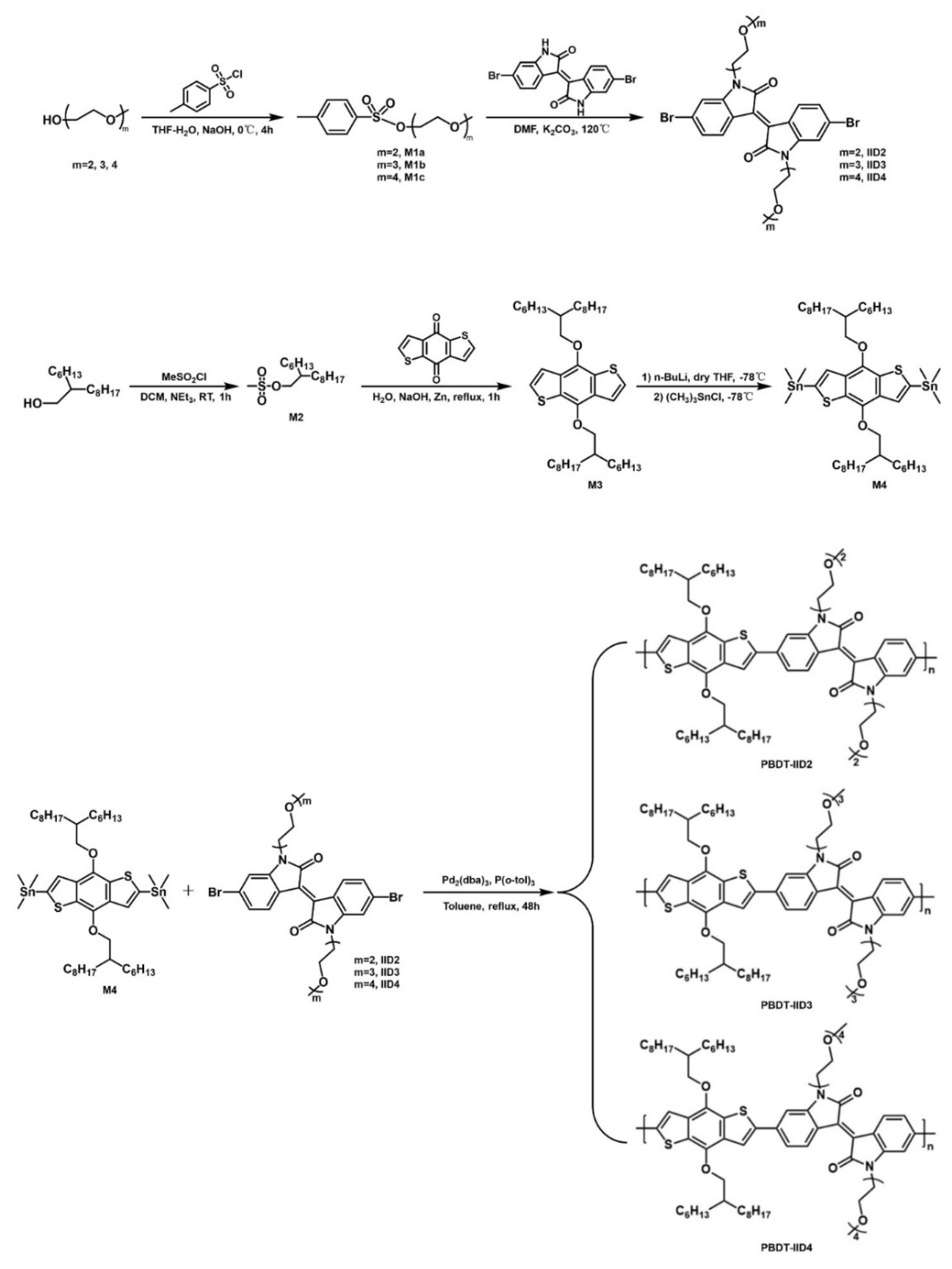
3.2. Synthesis
3.2.1. The Synthesis of Monomers M1a, M1b, and M1c
3.2.2. The Synthesis of Monomer M2
3.2.3. The Synthesis of Monomer M3
3.2.4. The Synthesis of Monomer M4
3.2.5. The Synthesis of Monomers IID2, IID3, and IID4
3.2.6. The Synthesis of Polymers PBDT-IID2, PBDT-IID3, and PBDT-IID4
4. Results and Discussion
4.1. Basic Characterization
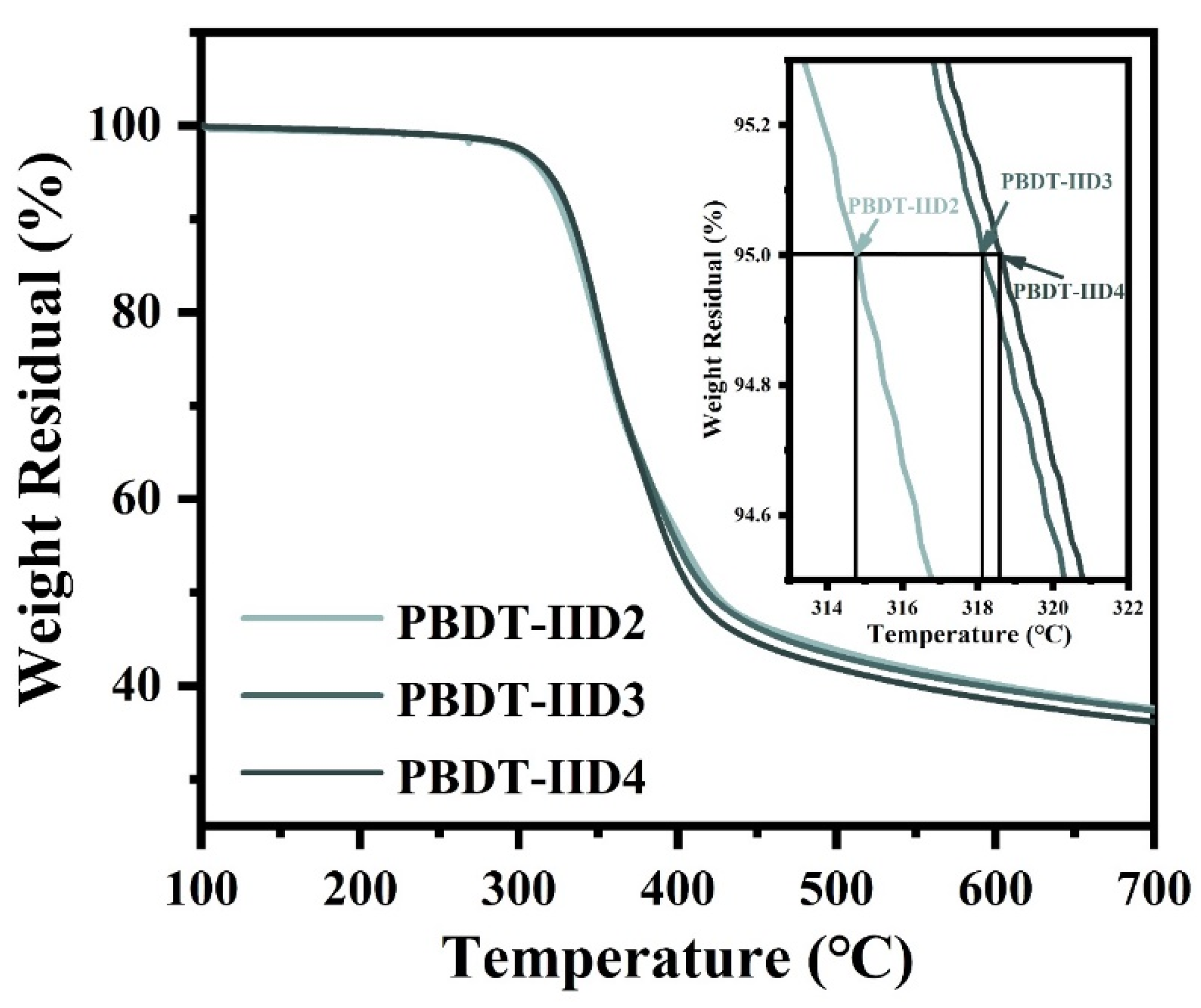
4.2. The Thermostability Properties of PBDT-IID Polymers
4.3. The Optical Properties of the PBDT-IID Polymers
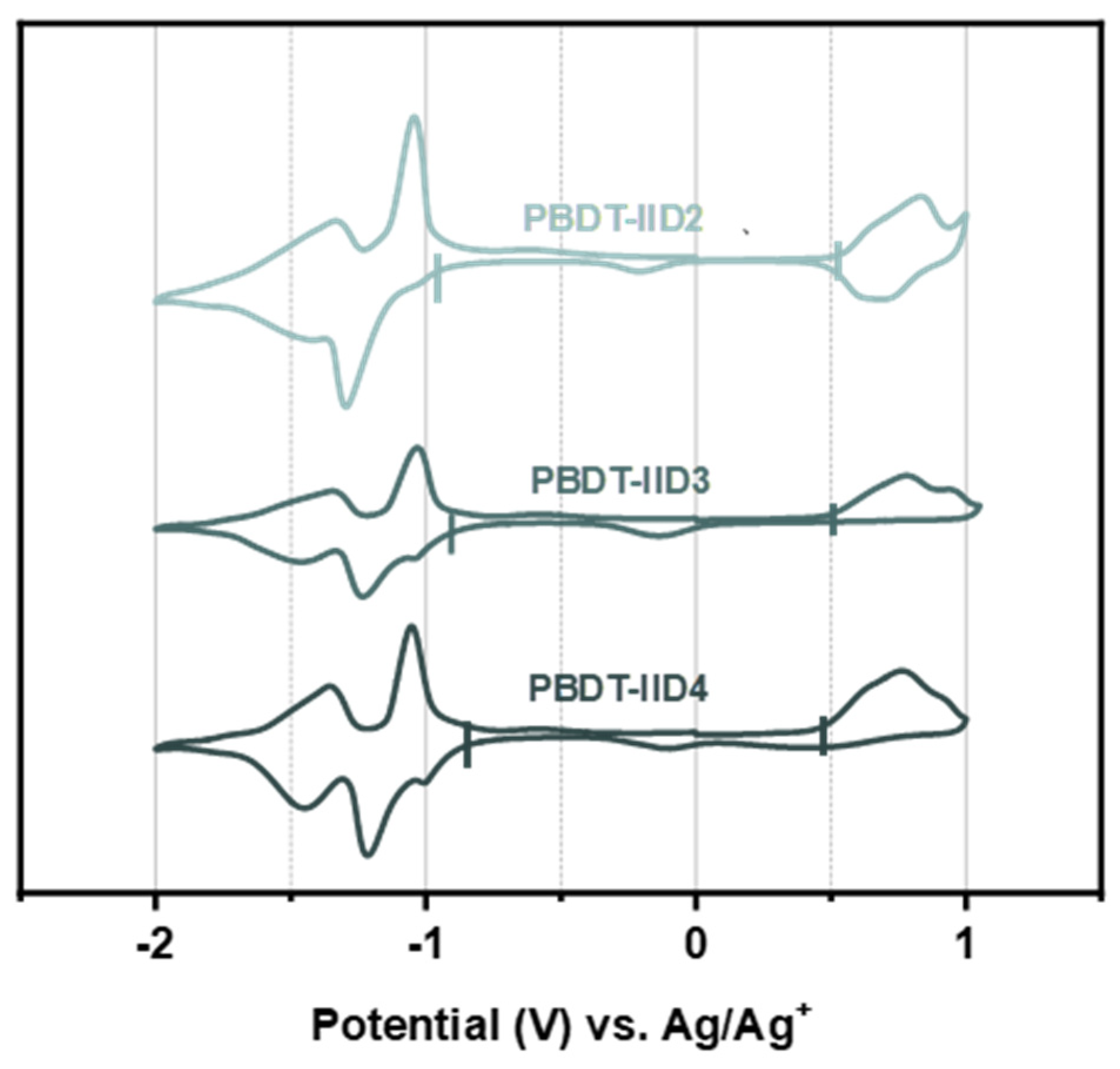
4.4. The Electrochemical Properties of the PBDT-IID Polymers
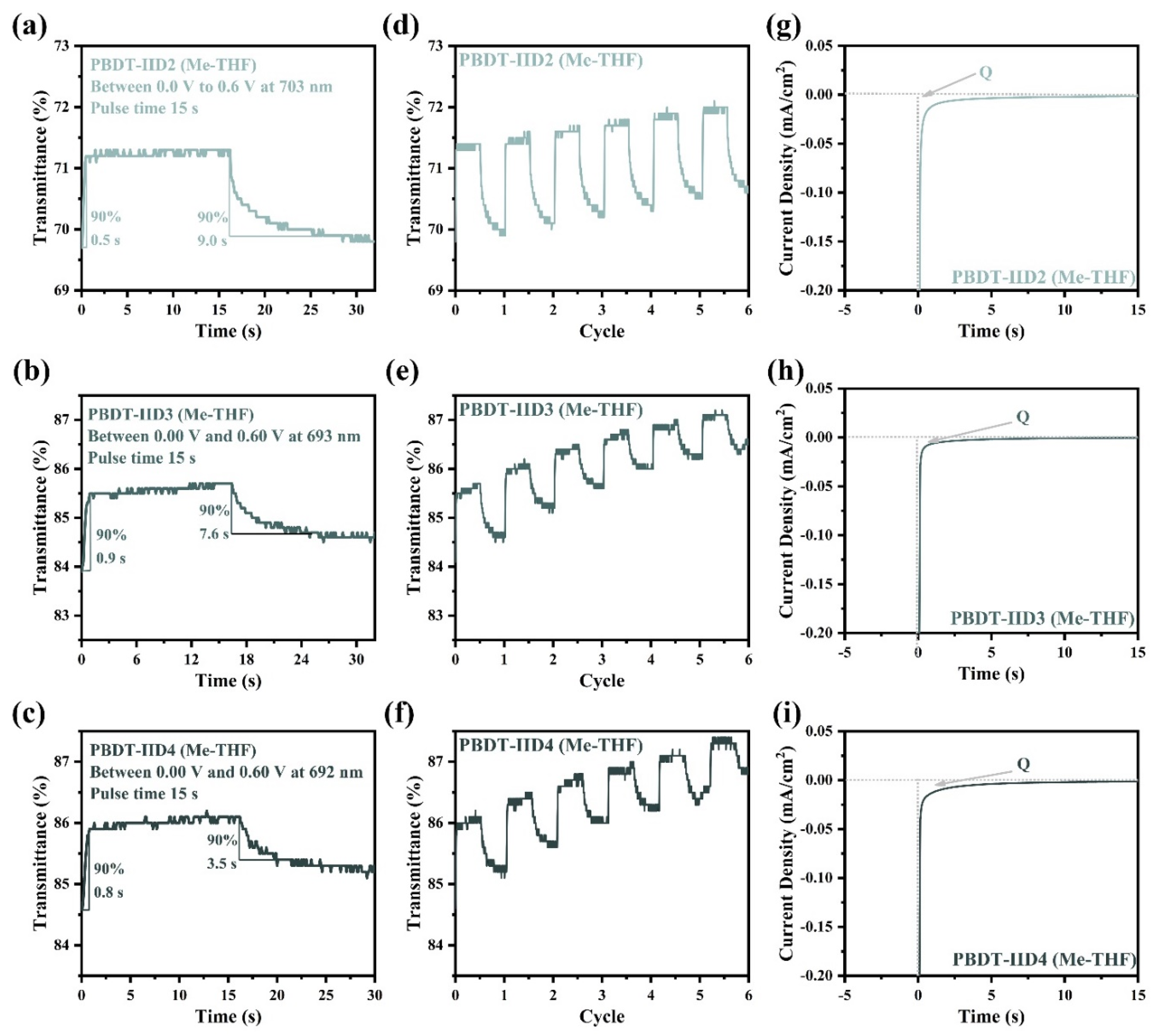
4.5. The Electrochromic Properties of the PBDT-IID Polymers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, Z.; Wang, X.; Cong, S.; Geng, F.; Zhao, Z. Fusing electrochromic technology with other advanced technologies: A new roadmap for future development. Mater. Sci. Eng. R Rep. 2020, 140, 100524. [Google Scholar] [CrossRef]
- Runnerstrom, E.L.; Llordes, A.; Lounis, S.D.; Milliron, D.J. Nanostructured electrochromic smart windows: Traditional materials and NIR-selective plasmonic nanocrystals. Chem. Commun. 2014, 50, 10555–10572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kline, W.M.; Lorenzini, R.G.; Sotzing, G.A. A review of organic electrochromic fabric devices. Color. Technol. 2014, 130, 73–80. [Google Scholar] [CrossRef]
- Eh, A.L.-S.; Tan, A.W.M.; Cheng, X.; Magdassi, S.; Lee, P.S. Recent advances in flexible electrochromic devices: Prerequisites, challenges, and prospects. Energy Technol. 2018, 6, 33–45. [Google Scholar] [CrossRef]
- Tong, Z.; Tian, Y.; Zhang, H.; Li, X.; Ji, J.; Qu, H.; Li, N.; Zhao, J.; Li, Y. Recent advances in multifunctional electrochromic energy storage devices and photoelectrochromic devices. Sci. China Chem. 2016, 60, 13–37. [Google Scholar] [CrossRef]
- Park, S.-I.; Quan, Y.-J.; Kim, S.-H.; Kim, H.; Kim, S.; Chun, D.-M.; Lee, C.S.; Taya, M.; Chu, W.-S.; Ahn, S.-H. A review on fabrication processes for electrochromic devices. Int. J. Precis. Eng. Manuf.-Green Technol. 2016, 3, 397–421. [Google Scholar] [CrossRef]
- Huang, Y.; Zhu, M.; Huang, Y.; Pei, Z.; Li, H.; Wang, Z.; Xue, Q.; Zhi, C. Multifunctional energy storage and conversion devices. Adv. Mater. 2016, 28, 8344–8364. [Google Scholar] [CrossRef]
- Granqvist, C.G.; Pehlivan, I.B.; Green, S.V.; LansaKer, P.C.; Niklasson, G.A. Oxide-based electrochromics: Advances in materials and devices. Mater. Res. Soc. Symp. Proc. 2011, 1328, 11–22. [Google Scholar] [CrossRef]
- Wolfart, F.; Hryniewicz, B.M.; Góes, M.S.; Corrêa, C.M.; Torresi, R.; Minadeo, M.A.O.S.; Córdoba de Torresi, S.I.; Oliveira, R.D.; Marchesi, L.F.; Vidotti, M. Conducting polymers revisited: Applications in energy, electrochromism and molecular recognition. J. Solid State Electrochem. 2017, 21, 2489–2515. [Google Scholar] [CrossRef]
- Lo, C.K.; Shen, D.E.; Reynolds, J.R. Fine-tuning the color hue of π-conjugated black-to-clear electrochromic random copolymers. Macromolecules 2019, 52, 6773–6779. [Google Scholar] [CrossRef]
- Schraff, S.; Sun, Y.; Pammer, F. Fulvenyl-functionalized polyisocyanides: Cross-conjugated electrochromic polymers with variable optical and electrochemical properties. Macromolecules 2018, 51, 5323–5335. [Google Scholar] [CrossRef]
- Lv, X.; Li, W.; Ouyang, M.; Zhang, Y.; Wright, D.S.; Zhang, C. Polymeric electrochromic materials with donor–acceptor structures. J. Mater. Chem. C 2017, 5, 12–28. [Google Scholar] [CrossRef]
- Chua, M.H.; Zhu, Q.; Tang, T.; Shah, K.W.; Xu, J. Diversity of electron acceptor groups in donor–acceptor type electrochromic conjugated polymers. Sol. Energy Mater. Sol. Cells 2019, 197, 32–75. [Google Scholar] [CrossRef]
- Li, X.; Perera, K.; He, J.; Gumyusenge, A.; Mei, J. Solution-processable electrochromic materials and devices: Roadblocks and strategies towards large-scale applications. J. Mater. Chem. C 2019, 7, 12761–12789. [Google Scholar] [CrossRef]
- Yu, H.; Shao, S.; Yan, L.; Meng, H.; He, Y.; Yao, C.; Xu, P.; Zhang, X.; Hu, W.; Huang, W. Side-chain engineering of green color electrochromic polymer materials: Toward adaptive camouflage application. J. Mater. Chem. C 2016, 4, 2269–2273. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Guo, Y.; Shi, J.; Yu, H.; Meng, H. Solution-processable neutral green electrochromic polymer containing thieno[3,2-b]thiophene derivative as unconventional donor units. Macromolecules 2016, 49, 7211–7219. [Google Scholar] [CrossRef]
- Us, C.N.; Icli Ozkut, M. Expanding the realm of soluble narrow band gap polymers with a benzobisthiadiazole derivative. Macromolecules 2016, 49, 3009–3015. [Google Scholar] [CrossRef]
- Stalder, R.; Mei, J.; Subbiah, J.; Grand, C.; Estrada, L.A.; So, F.; Reynolds, J.R. N-type conjugated polyisoindigos. Macromolecules 2011, 44, 6303–6310. [Google Scholar] [CrossRef]
- Stalder, R.; Mei, J.; Graham, K.R.; Estrada, L.A.; Reynolds, J.R. Isoindigo, a versatile electron-deficient unit for high-performance organic ectronics. Chem. Mater. 2013, 26, 664–678. [Google Scholar] [CrossRef]
- Yao, H.; Ye, L.; Zhang, H.; Li, S.; Zhang, S.; Hou, J. Molecular design of benzodithiophene-based organic photovoltaic materials. Chem. Rev. 2016, 116, 7397–7457. [Google Scholar] [CrossRef]
- Yang, S.F.; Liu, Z.T.; Cai, Z.X.; Dyson, M.J.; Stingelin, N.; Chen, W.; Ju, H.J.; Zhang, G.X.; Zhang, D.Q. Diketopyrrolopyrrole-based conjugated polymer entailing triethylene glycols as side chains with high thin-film charge mobility without post-treatments. Adv. Sci. 2017, 4, 1700048. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Chen, Z.; Wei, W.; Jiang, Z. Fluorinated benzothiadiazole-based conjugated polymers for high-performance polymer solar cells without any processing additives or post-treatments. J. Am. Chem. Soc. 2013, 135, 17060–17068. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yan, L.; Rech, J.J.; Hu, J.; Zhang, Q.; You, W. Green-solvent-processed conjugated polymers for organic solar cells: The impact of oligoethylene glycol side chains. ACS Appl. Polym. Mater. 2019, 1, 804–814. [Google Scholar] [CrossRef]
- Shchegolkov, A.V.; Jang, S.-H.; Shchegolkov, A.V.; Rodionov, Y.V.; Sukhova, A.O.; Lipkin, M.S. A brief overview of electrochromic materials and related devices: A nanostructured materials perspective. Nanomaterials 2021, 11, 2376. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Song, H.; Chen, X.; Xie, Z.; Liu, J.; Wang, L. Replacing alkyl with oligo(ethylene glycol) as side chains of conjugated polymers for close π–π stacking. Macromolecules 2015, 48, 4357–4363. [Google Scholar] [CrossRef]
- Steyrleuthner, R.; Schubert, M.; Howard, I.; Klaumunzer, B.; Schilling, K.; Chen, Z.; Saalfrank, P.; Laquai, F.; Facchetti, A.; Neher, D. Aggregation in a high-mobility n-type low-bandgap copolymer with implications on semicrystalline morphology. J. Am. Chem. Soc. 2012, 134, 18303–18317. [Google Scholar] [CrossRef]
- Gu, X.; Shaw, L.; Gu, K.; Toney, M.F.; Bao, Z. The meniscus-guided deposition of semiconducting polymers. Nat. Commun. 2018, 9, 534. [Google Scholar] [CrossRef] [Green Version]
- Nahid, M.M.; Welford, A.; Gann, E.; Thomsen, L.; Sharma, K.P.; McNeill, C.R. Nature and extent of solution aggregation determines the performance of P(NDI2OD-T2) thin-film transistors. Adv. Electron. Mater. 2018, 4, 1700559. [Google Scholar] [CrossRef]
- Hu, Z.; Wang, Z.; Zhang, X.; Tang, H.; Liu, X.; Huang, F.; Cao, Y. Conjugated polymers with oligoethylene glycol side chains for improved photocatalytic hydrogen evolution. iScience 2019, 13, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Lei, T.; Dou, J.H.; Pei, J. Influence of alkyl chain branching positions on the hole mobilities of polymer thin-film transistors. Adv. Mater. 2012, 24, 6457–6461. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Z.; Ding, Z.; Liu, J.; Wang, L. Diketopyrrolopyrrole-based conjugated polymers bearing branched oligo(ethylene glycol) side chains for photovoltaic devices. Angew. Chem. Int. Ed. 2016, 55, 10376–10380. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Liu, W.; Ling, Y.; Tang, H. Synthesis and thermoresponsive properties of OEGylated polypeptide with a LCST at body temperature in water and with a UCST in alcohol or ethanol/water solvent mixture. J. Polym. Sci. A Polym. Chem. 2018, 56, 163–173. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, P.; Chen, Y.; Luo, H.; Zhang, Z.; Wang, H.; Li, X.; Yu, G.; Li, Y. Thieno[3,2-b]thiophene-bridged D−π–A polymer semiconductor based on benzo[1,2-b:4,5-b′]dithiophene and benzoxadiazole. Macromolecules 2013, 46, 4805–4812. [Google Scholar] [CrossRef]
- Skotheim, T.A.; Reynolds, J. Conjugated Polymers, Theory, Synthesis, Properties, and Characterization; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Huang, D.-C.; Wu, J.-T.; Fan, Y.-Z.; Liou, G.-S. Preparation and optoelectronic behaviours of novel electrochromic devices based on triphenylamine-containing ambipolar materials. J. Mater. Chem. C 2017, 5, 9370–9375. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Liou, G.-S. Poly(triphenylamine)s derived from oxidative coupling reaction: Substituent effects on the polymerization, electrochemical, and electro-optical properties. J. Polym. Sci. A Polym. Chem. 2009, 47, 285–294. [Google Scholar] [CrossRef]
- Qi, S.; Wang, C.; Liu, Z.; Han, Y.; Bai, F.; Chen, Z. Electrochromic and photovoltaic properties of benzothiadiazole-based donor-acceptor conjugated polymers with oligo(ethylene glycol) side chains. Dyes Pigment. 2022, 204, 110432. [Google Scholar] [CrossRef]
- Kar, P. Doping in Conjugated Polymers; Scrivener Publishing: Beverly, MA, USA, 2013. [Google Scholar]
- Reynolds, J.R.; Thompson, B.C.; Skotheim, T.A. Conjugated Polymers: Perspective, Theory, and New Materials; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Fu, H.; Yan, S.; Yang, T.; Yin, M.; Zhang, L.; Shao, X.; Dong, Y.; Li, W.; Zhang, C. New dual conjugated polymer electrochromic device with remarkable yellow-to-green switch for adaptive camouflage. Chem. Eng. J. 2022, 438, 135455. [Google Scholar] [CrossRef]
- Li, H.; Yang, T.; Li, L.; Lv, S.; Li, S. A camouflaged film imitating the chameleon skin with color-changing microfluidic systems based on the color information identification of background. J. Bionic Eng. 2021, 18, 1137–1146. [Google Scholar] [CrossRef]
- Koyuncu, S.; Koyuncu, F.B. A new ITO-compatible side chain-functionalized multielectrochromic polymer for use in adaptive camouflage-like electrochromic devices. React. Funct. Polym. 2018, 131, 174–180. [Google Scholar] [CrossRef]
- Meng, J.; Li, X.; Qin, M.; Pei, Y.; Yang, S.; Lan, Y.; Wang, R.; Chen, G. Effects of pore size of reverse opal structured PEDOT films on their electrochromic performances. Org. Electron. 2017, 50, 16–24. [Google Scholar] [CrossRef]
- Zhuang, B.; Wang, X.; Zhang, Q.; Liu, J.; Jin, Y.; Wang, H. Nanoengineering of poly(3,4-ethylenedioxythiophene) for boosting electrochemical applications. Sol. Energy Mater. Sol. Cells 2021, 232, 111357. [Google Scholar] [CrossRef]
- Beaujuge, P.M.; Reynolds, J.R. Color control in π-conjugated organic polymers for use in electrochromic devices. Chem. Rev. 2010, 110, 268–320. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | GPC (kDa) [a] | Thermal Stabilities (°C) | ||
|---|---|---|---|---|
| Mn | Mw | Đ | Td5% [b] | |
| PBDT-IID2 | 54 | 118.8 | 2.20 | 314.7 |
| PBDT-IID3 | 49 | 179.8 | 3.67 | 318.0 |
| PBDT-IID4 | 37 | 181.3 | 4.91 | 318.5 |
| Polymer | PBDT-IID2 | PBDT-IID3 | PBDT-IID4 | |
|---|---|---|---|---|
| Solution [a] | BDT π-π* | 380 | 378 | 377 |
| IID π-π* | 450 | 445 | 444 | |
| λmax (nm) | 687 | 672 | 669 | |
| Film [b] | BDT π-π* | 391 | 388 | 385 |
| IID π-π* | 452 | 447 | 445 | |
| λmax (nm) | 702 | 696 | 692 | |
| λonset (nm) | 807 | 793 | 787 | |
| (eV) | 1.54 | 1.56 | 1.58 | |
| Polymer | Oxidation (V) | Reduction (V) | Energy Level (eV) | |||||
|---|---|---|---|---|---|---|---|---|
| HOMO | LUMO | |||||||
| PBDT-IID2 | 0.52 | 0.84 | −1.04 | −0.88 | 0.67 | −1.30 | 5.32 | 3.78 |
| PBDT-IID3 | 0.49 | 0.78 | −1.03 | −0.84 | -- | −1.24 | 5.29 | 3.73 |
| PBDT-IID4 | 0.45 | 0.76 | −1.04 | −0.82 | -- | −1.21 | 5.25 | 3.67 |
| Solvents | Polymers | ΔT (%) | Response Time (s) tc/tb | CE (cm2/C) |
|---|---|---|---|---|
| Me-THF | PBDT-IID2 | 1.53 | 0.5 s/9.0 s | 213.10 |
| PBDT-IID3 | 1.46 | 0.9 s/7.6 s | 214.12 | |
| PBDT-IID4 | 1.51 | 0.8 s/3.5 s | 242.06 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Zhai, Y.; Chao, D.; Chen, Z.; Jiang, Z. Preparation and Electrochromic Properties of Benzodithiophene-Isoindigo Conjugated Polymers with Oligoethylene Glycol Side Chains. Materials 2023, 16, 60. https://doi.org/10.3390/ma16010060
Wang Q, Zhai Y, Chao D, Chen Z, Jiang Z. Preparation and Electrochromic Properties of Benzodithiophene-Isoindigo Conjugated Polymers with Oligoethylene Glycol Side Chains. Materials. 2023; 16(1):60. https://doi.org/10.3390/ma16010060
Chicago/Turabian StyleWang, Qilin, Yuehui Zhai, Danming Chao, Zheng Chen, and Zhenhua Jiang. 2023. "Preparation and Electrochromic Properties of Benzodithiophene-Isoindigo Conjugated Polymers with Oligoethylene Glycol Side Chains" Materials 16, no. 1: 60. https://doi.org/10.3390/ma16010060