
[2.2]Paracyclophane Derivatives as Building Blocks for Coordination Polymers
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. General Procedure A—Suzuki Coupling; Compound 8
2.1.2. Compound 5
2.1.3. Compound 6
2.1.4. Compound 7
2.1.5. Compound 14
2.1.6. Compound 17
2.1.7. General Procedure B—Ester Hydrolysis; Compound 12
2.1.8. Compound 9
2.1.9. Compound 10
2.1.10. Compound 11
2.1.11. Compound 15
2.1.12. Compound 18
2.1.13. Compound 19—2D Zinc Coordination Polymer
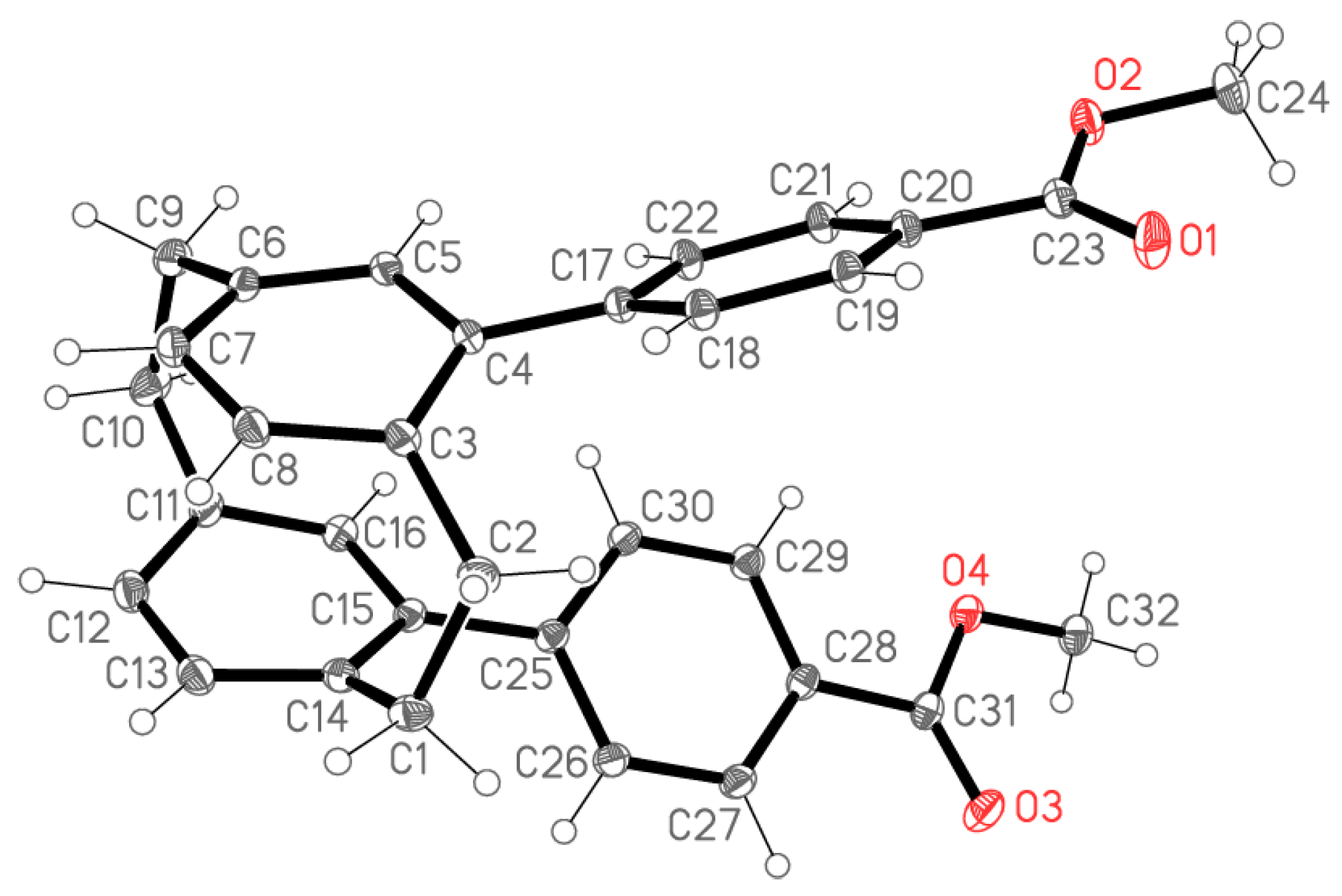
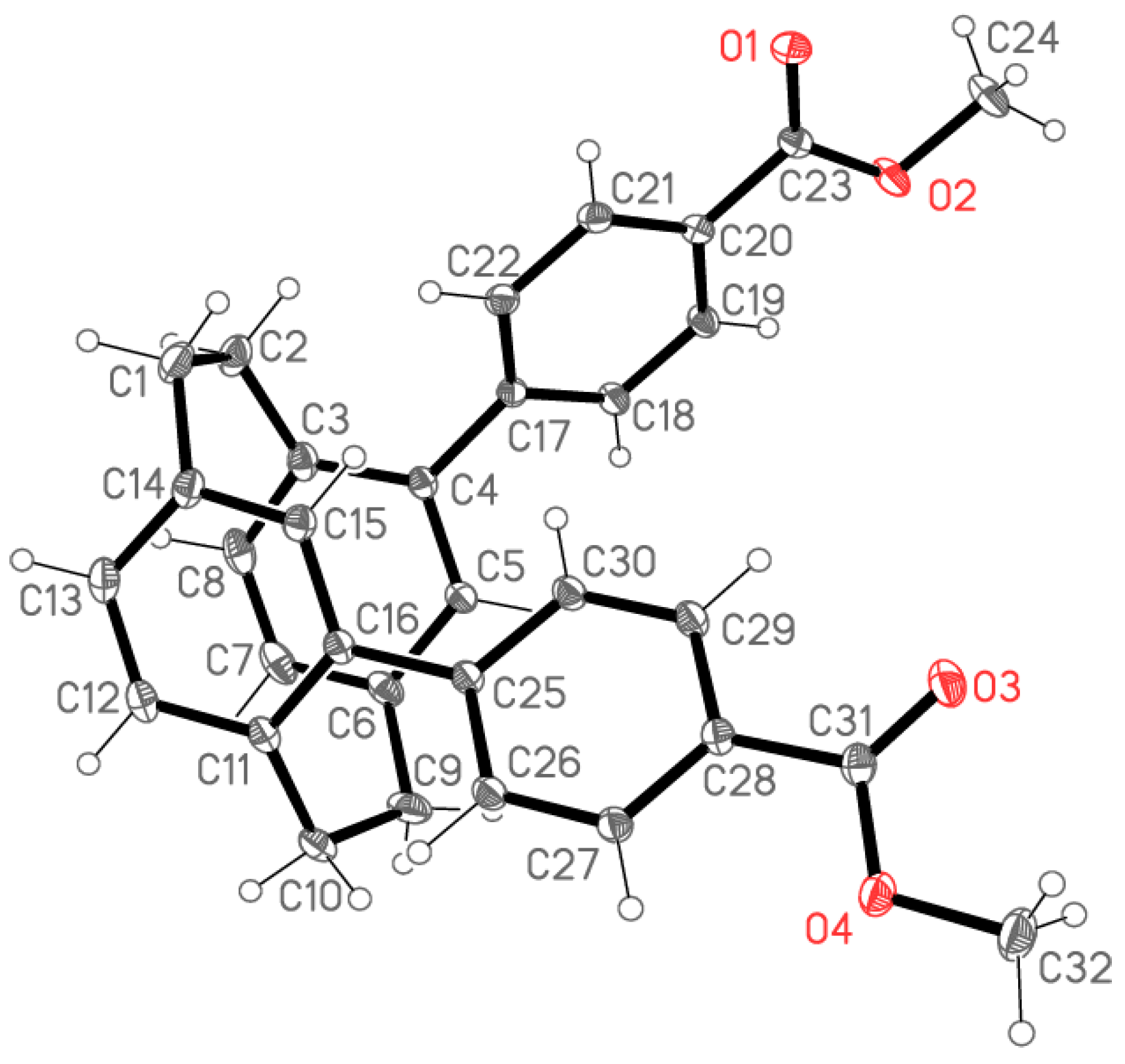
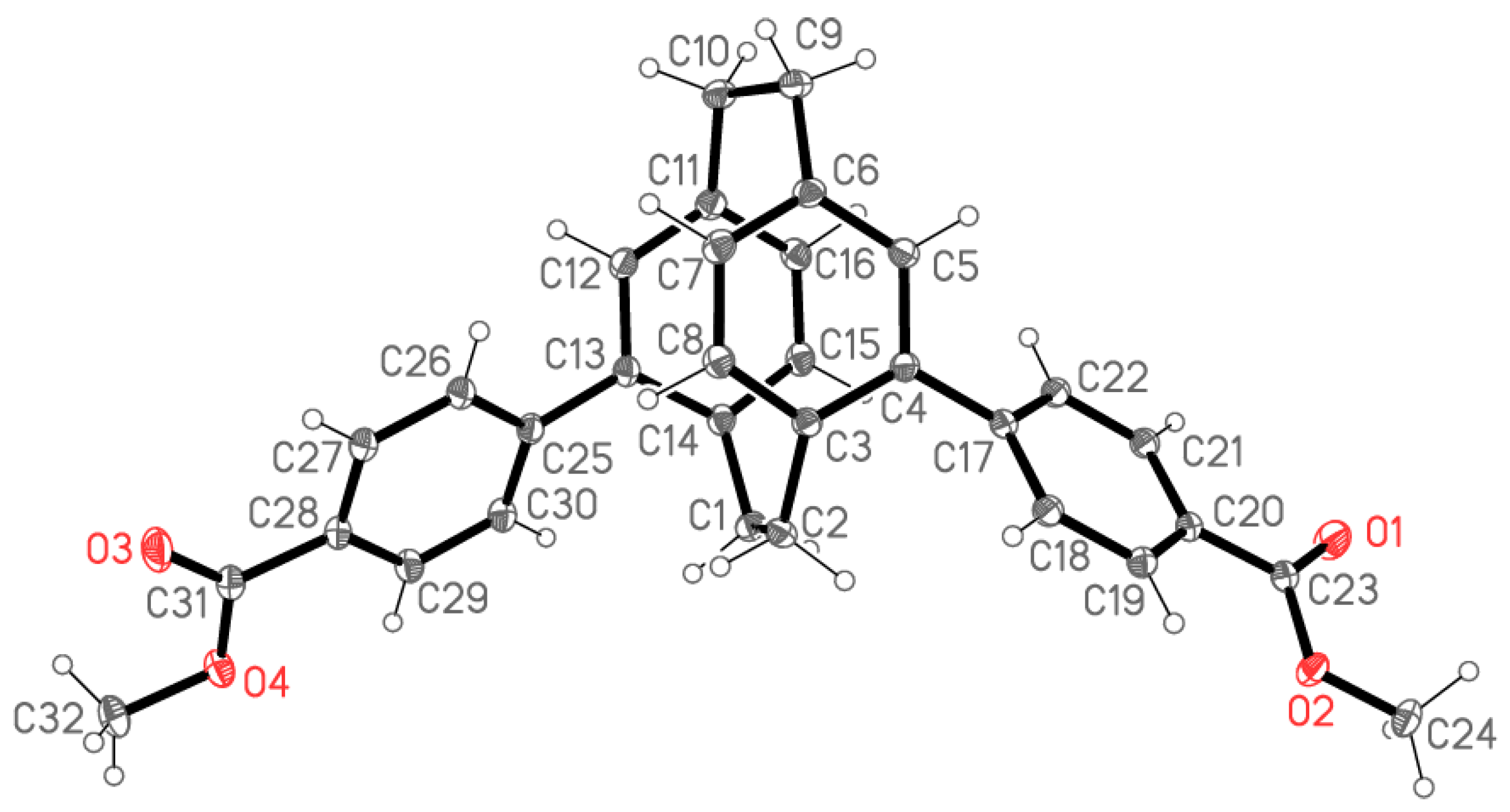
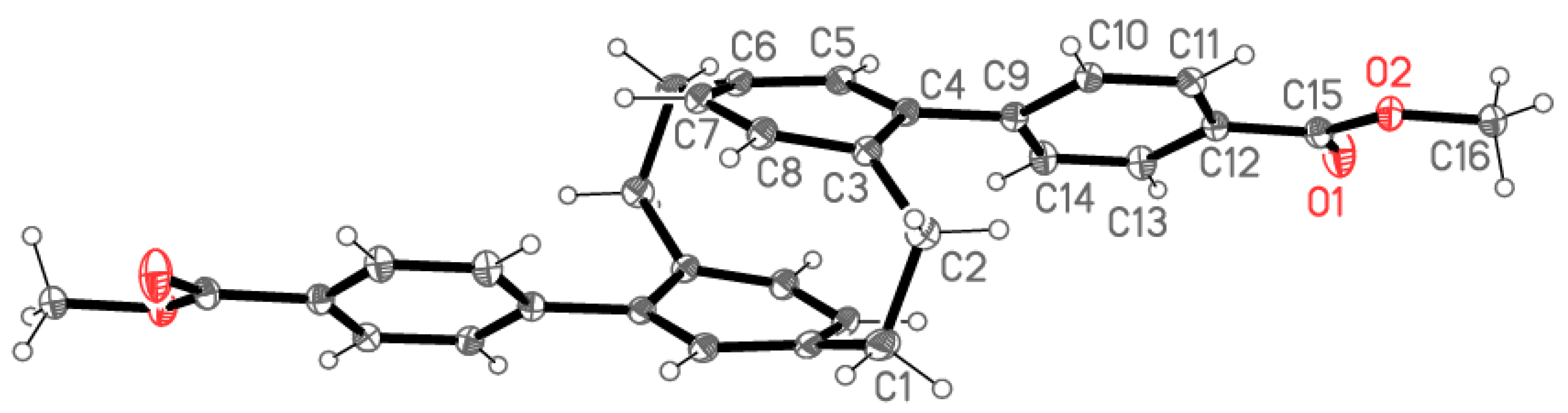
2.2. X-ray Crystallography
Special Features and Exceptions
3. Results and Discussion
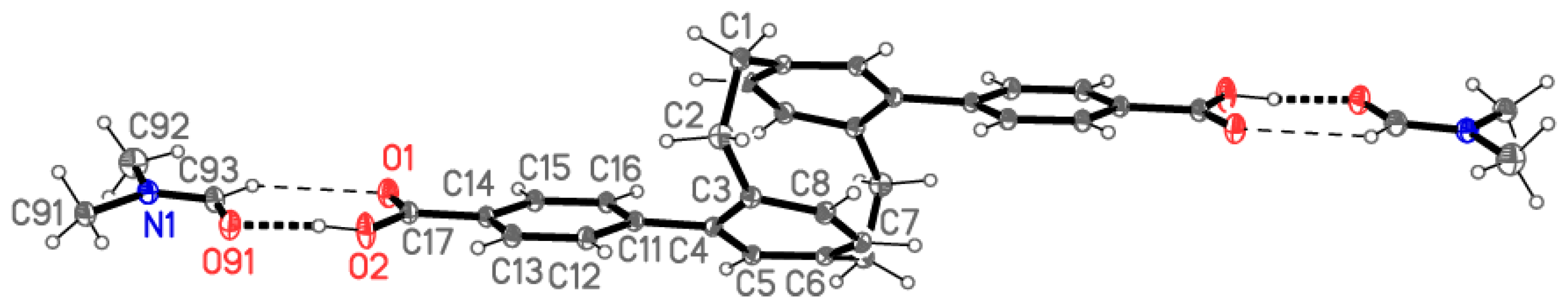
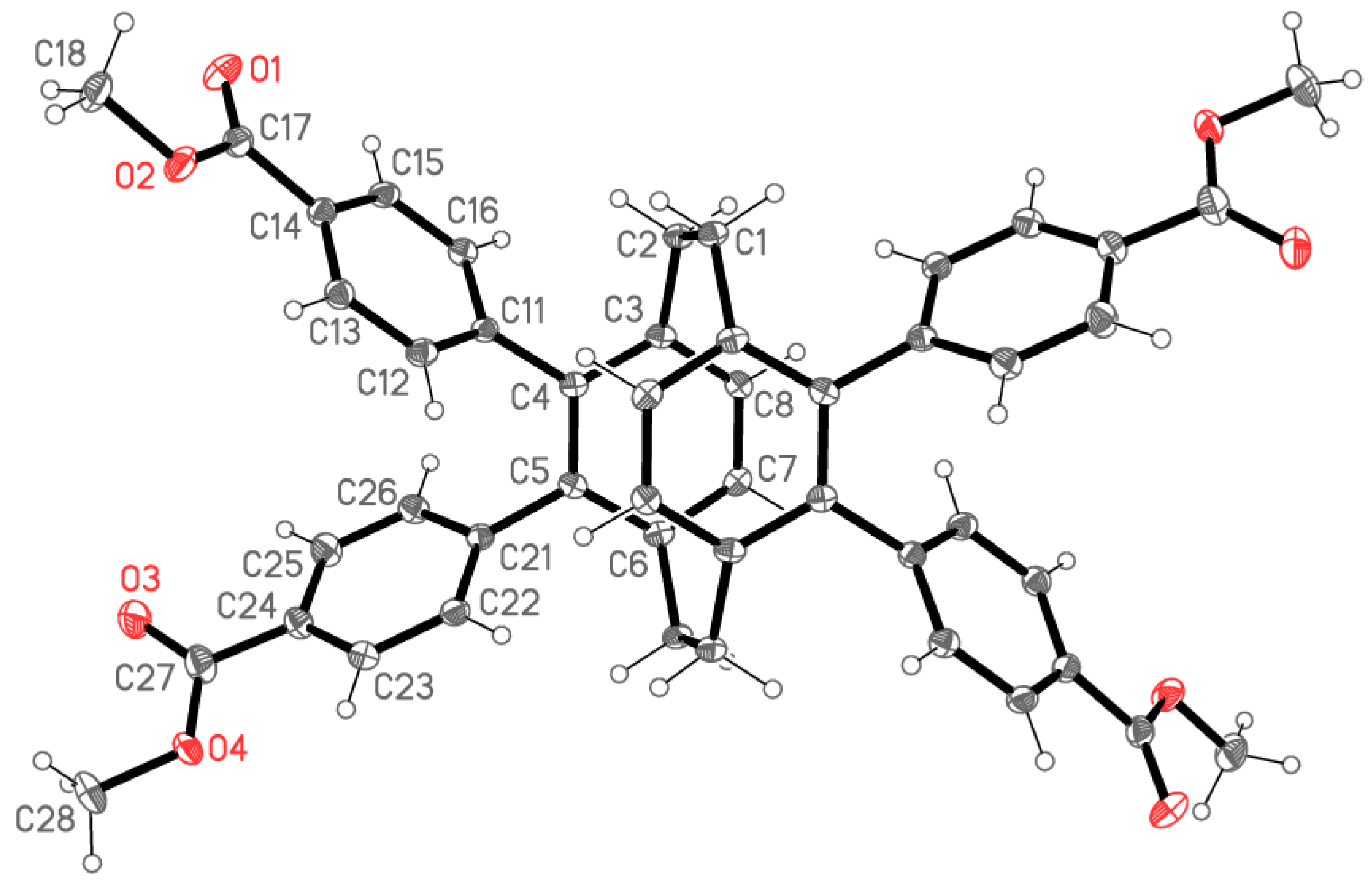
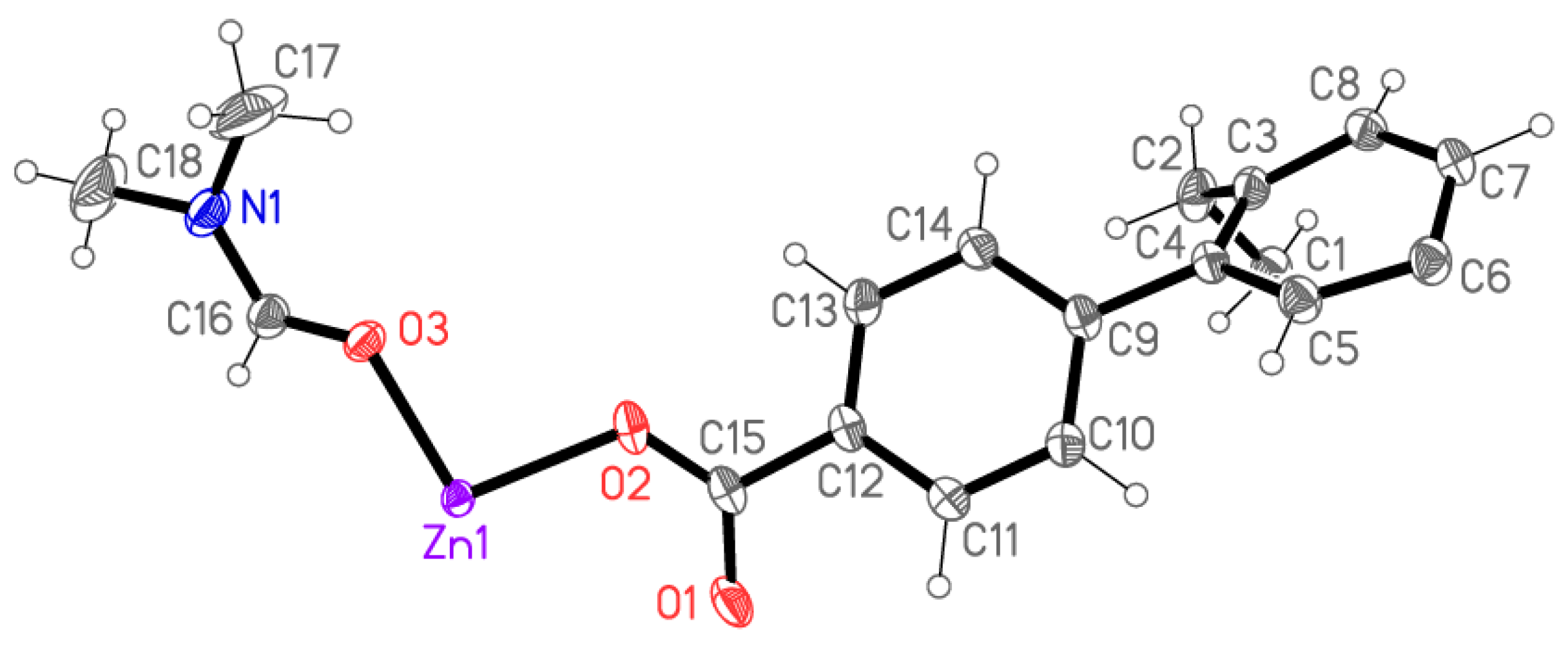
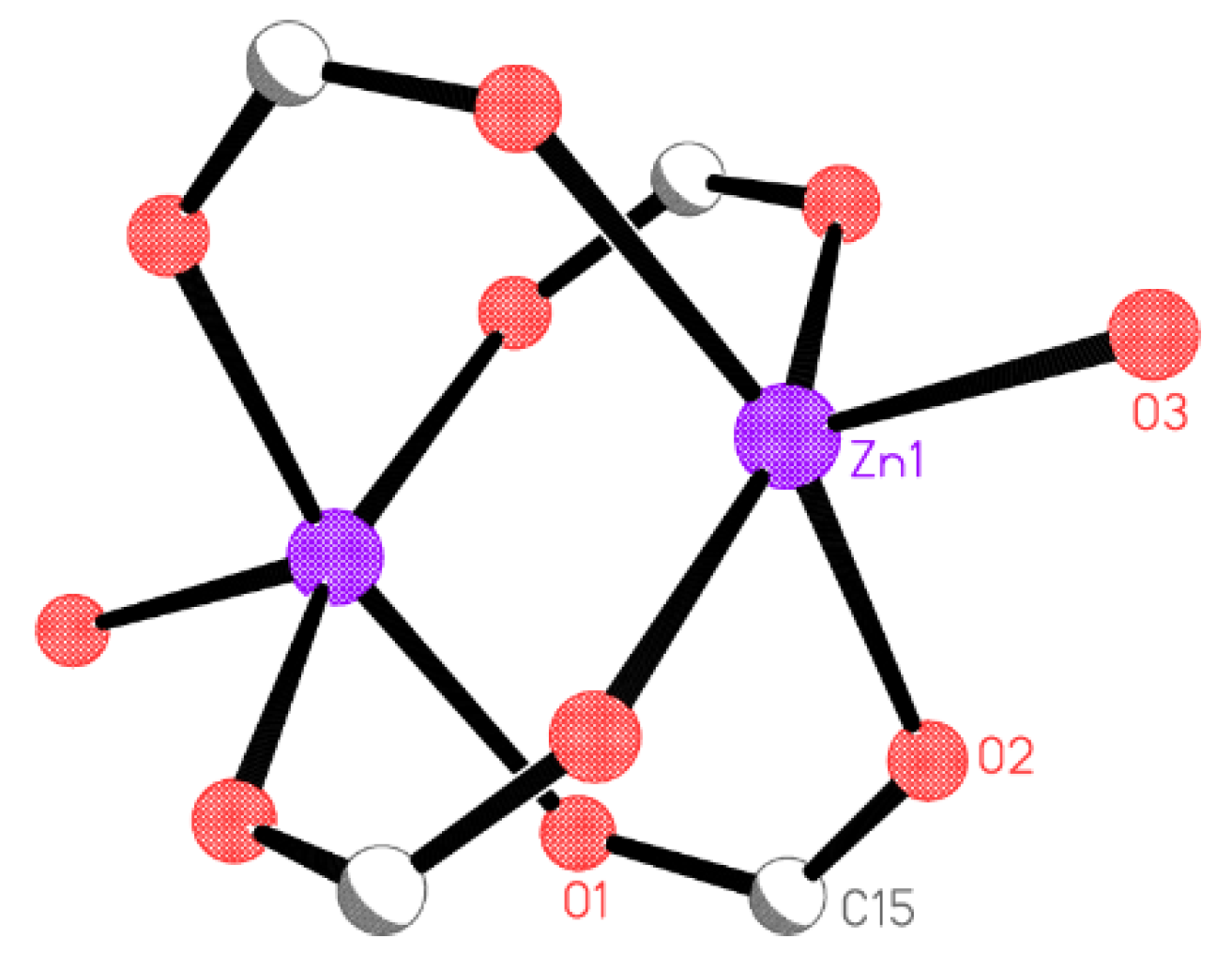
X-ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, C.J.; Farthing, A.C. Preparation and Structure of Di-p-Xylylene. Nature 1949, 164, 915–916. [Google Scholar] [CrossRef]
- Boyd, R.H. The heat of combustion and strain energy of 2,2-paracyclophane. Tetrahedron 1966, 22, 119–122. [Google Scholar] [CrossRef]
- Vögtle, F. Cyclophane Chemistry; Wiley: Chichester, UK, 1993. [Google Scholar]
- Vorontsova, N.V.; Rozenberg, V.I.; Sergeeva, E.V.; Vorontsov, E.V.; Starikova, Z.A.; Lyssenko, K.A.; Hopf, H. Symmetrically Tetrasubstituted [2.2]Paracyclophanes: Their Systematization and Regioselective Synthesis of Several Types of Bis-Bifunctional Derivatives by Double Electrophilic Substitution. Chem. Eur. J. 2008, 14, 4600–4617. [Google Scholar] [CrossRef] [PubMed]
- Hassan, Z.; Spuling, E.; Knoll, D.M.; Bräse, S. Regioselective Functionalization of [2.2]Paracyclophanes: Recent Synthetic Progress and Perspectives. Angew. Chem. Int. Ed. 2020, 59, 2156–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopf, H. [2.2]Paracyclophanes in Polymer Chemistry and Materials Science. Angew. Chem. Int. Ed. 2008, 47, 9808–9812. [Google Scholar] [CrossRef]
- Sugiura, K. [2.2]Paracyclophane-Based Chiral Platforms for Circularly Polarized Luminescence Fluorophores and Their Chiroptical Properties: Past and Future. Front. Chem. 2020, 8, 700. [Google Scholar] [CrossRef]
- Spuling, E.; Sharma, N.; Samuel, I.D.W.; Zysman-Colman, E.; Bräse, S. (Deep) blue through-space conjugated TADF emitters based on [2.2]paracyclophanes. Chem. Commun. 2018, 54, 9278–9281. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.H.; Ding, Y.X.; Wua, B.; Zhou, Y.G. Design and synthesis of chiral and regenerable [2.2]paracyclophane-based NAD(P)H models and application in biomimetic reduction of flavonoids. Chem. Sci. 2020, 11, 10220–10224. [Google Scholar] [CrossRef]
- Gong, W.; Xie, H.; Idrees, K.B.; Son, F.A.; Chen, Z.; Sha, F.; Liu, Y.; Cui, Y.; Farha, O.K. Water Sorption Evolution Enabled by Reticular Construction of Zirconium Metal–Organic Frameworks Based on a Unique [2.2]Paracyclophane Scaffold. J. Am. Chem. Soc. 2022, 144, 1826–1834. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, W.; Hou, B.; Liu, Y.; Cui, Y. Planar Chiral [2.2]Paracyclophane-Based Zr(IV) Metal–Organic Frameworks. CCS Chem. 2022. [Google Scholar] [CrossRef]
- Kuroda-Sowa, T.; Liu, S.Q.; Yamazaki, Y.; Munakata, M.; Maekawa, M.; Suenaga, Y.; Konaka, H.; Nakagawa, H. Silver(I) Compounds Consisting of [2.2]Paracyclophane: Reversible Guest-Driven Solid-State Transformation and Incorporation Behavior. Inorg. Chem. 2005, 44, 1686–1692. [Google Scholar] [CrossRef]
- Liu, S.Q.; Konaka, H.; Kuroda-Sowa, T.; Maekawa, M.; Suenaga, Y.; Ning, G.L.; Munakata, M. 3D coordination polymers of [2.2]paracyclophane and in situ silver(I) perfluoro-dicarboxylates: Effects of the dicarboxylate spacers and conformations on the formation of complexes. Inorg. Chim. Acta 2005, 358, 919–926. [Google Scholar] [CrossRef]
- Munakata, M.; Wu, L.P.; Ning, G.L.; Kuroda-Sowa, T.; Maekawa, M.; Suenaga, Y.; Maeno, N. Construction of Metal Sandwich Systems Derived from Assembly of Silver(I) Complexes with Polycyclic Aromatic Compounds. J. Am. Chem. Soc. 1999, 121, 4968–4976. [Google Scholar] [CrossRef]
- Mackenzie, C.F.R.; Delforce, L.; Martir, D.R.; Cordes, D.B.; Slawin, A.M.Z.; Zysman-Colman, E. A Luminescent 1D Silver Polymer Containing [2.2]Paracyclophane Ligands. Front. Chem. 2021, 9, 728845. [Google Scholar] [CrossRef]
- Papaefstathiou, G.S.; Friscic, T.; MacGillivray, L.R. Design and Construction of a 2D Metal Organic Framework with Multiple Cavities: A Nonregular Net with a Paracyclophane that Codes for Multiply Fused Nodes. J. Am. Chem. Soc. 2005, 127, 14160–14161. [Google Scholar] [CrossRef]
- Xue, X.; Wang, J.; Zhu, Q.; Xue, Y.; Liu, H. A two-year water-stable 2D MOF with aqueous NIR photothermal conversion ability. Dalton Trans. 2021, 50, 1374–1383. [Google Scholar] [CrossRef]
- Bejan, D.; Bahrin, L.G.; Shova, S.; Marangoci, N.L.; Kökҫam-Demir, Ü.; Lozan, V.; Janiak, C. New Microporous Lanthanide Organic Frameworks. Synthesis, Structure, Luminescence, Sorption, and Catalytic Acylation of 2-Naphthol. Molecules 2020, 25, 3055. [Google Scholar] [CrossRef]
- Bahrin, L.G.; Bejan, D.; Shova, S.; Gdaniec, M.; Fronc, M.; Lozan, V.; Janiak, C. Alkali- and alkaline-earth metal–organic networks based on a tetra(4-carboxyphenyl)bimesitylene-linker. Polyhedron 2019, 173, 114128. [Google Scholar] [CrossRef]
- Bejan, D.; Bahrin, L.G.; Cojocaru, C.; Trandabat, A.F.; Marangoci, N.L.; Rotaru, A.; Shova, S. The use of C1 symmetry imidazole-carboxylate building block and auxiliary acetate co-ligand for assembly of a 2D wave-like zinc(II) coordination polymer: Experimental and theoretical study. J. Coord. Chem. 2020, 73, 2250–2264. [Google Scholar] [CrossRef]
- Bahrin, L.G.; Nicolescu, A.; Shova, S.; Marangoci, N.L.; Birsa, L.M.; Sarbu, L.G. Nitrogen-Based Linkers with a Mesitylene Core: Synthesis and Characterization. Molecules 2021, 26, 5952. [Google Scholar] [CrossRef]
- Bejan, D.; Dascalu, I.A.; Shova, S.; Trandabat, A.F.; Bahrin, L.G. Mesitylene Tribenzoic Acid as a Linker for Novel Zn/Cd Metal-Organic Frameworks. Materials 2022, 15, 4247. [Google Scholar] [CrossRef] [PubMed]
- Bahrin, L.G.; Sarbu, L.G.; Jones, P.G.; Birsa, L.M.; Hopf, H. [2.2]Paracyclophane-Bis(triazole) Systems: Synthesis and Photochemical Behavior. Chem. Eur. J. 2017, 23, 12338–12345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahrin, L.G.; Hopf, H.; Jones, P.G.; Birsa, M.L.; Sarbu, L.G. An Approach to Paracyclophane-Based Tetrathiafulvalenes: Synthesis and Characterization of a Pseudo-Geminal [2.2]Paracyclophane 1,3-Dithia-2-Thione. Molecules 2020, 25, 5262. [Google Scholar] [CrossRef] [PubMed]
- Sarbu, L.G.; Bahrin, L.G.; Hopf, H.; Birsa, M.L. Chalchogenide Induced Intramolecular Interactions In [2.2]Paracyclophanes: A Review. Stud. Chem. 2019, 3, 7–16. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Clausen, H.F.; Poulen, R.D.; Bond, A.D.; Chevallier, M.A.S.; Iversen, B.B. Solvothermal synthesis of new metal organic framework structures in the zinc–terephthalic acid–dimethyl formamide system. J. Solid State Chem. 2005, 178, 3342–3351. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 5 | 6 | 7 | 8 |
|---|---|---|---|---|
| CCDC number | 2245482 | 2245483 | 2245484 | 2245485 |
| Formula | C32H28O4 | C32H28O4 | C32H28O4 | C32H28O4 |
| Mr | 476.54 | 476.54 | 476.54 | 476.54 |
| Crystal size (mm) | 0.2 × 0.2 × 0.04 | 0.2 × 0.15 × 0.15 | 0.22 × 0.17 × 0.17 | 0.2 × 0.2 × 0.05 |
| Crystal system | orthorhombic | triclinic | triclinic | triclinic |
| Space group | Pca21 | P(−1) | P(−1) | P(−1) |
| Temperature (°C) | −173 | −173 | −173 | −173 |
| a (Å) | 26.8431(5) | 10.1243(2) | 6.9793(2) | 6.88619(13) |
| b (Å) | 7.86457(15) | 10.3597(3) | 9.3525(3) | 8.94644(16) |
| c (Å) | 11.1615(2) | 12.7502(3) | 18.0147(6) | 9.47904(18) |
| α (°) | 90 | 67.824(2) | 92.488(2) | 92.4255(14) |
| β (°) | 90 | 75.462(2) | 91.462(2) | 94.0838(16) |
| γ (°) | 90 | 81.498(2) | 93.942(2) | 92.4901(14) |
| V (Å3) | 2356.29 | 1196.62 | 1171.52 | 581.34 |
| Z | 4 | 2 | 2 | 1 |
| Dx (Mg m−3) | 1.343 | 1.323 | 1.351 | 1.361 |
| λ (Å) | 0.71073 (Mo Kα) | 0.71073 (Mo Kα) | 0.71073 (Mo Kα) | 0.71073 (Mo Kα) |
| μ (mm−1) | 0.09 | 0.09 | 0.09 | 0.09 |
| Transmissions | 0.808–1.000 | 0.927–1.000 | 0.982–1.000 | 0.801–1.000 |
| F(000) | 1008 | 504 | 504 | 252 |
| 2θmax | 78.7 | 82.6 | 76.7 | 82.7 |
| Refl. measured | 175,596 | 153,237 | 17,398 | 74,728 |
| Refl. indep. | 13,671 | 15,664 | 17,398 | 7643 |
| Rint | 0.044 | 0.029 | n/a | 0.25 |
| Parameters | 351 | 351 | 352 | 176 |
| Restraints | 16 | 15 | 15 | 3 |
| wR(F2, all refl.) | 0.092 | 0.117 | 0.131 | 0.123 |
| R(F, >4σ(F)) | 0.034 | 0.037 | 0.045 | 0.039 |
| S | 1.06 | 1.06 | 1.10 | 1.04 |
| Max. Δp (e Å−3) | 0.48, −0.20 | 0.59, −0.28 | 0.53, −0.24 | 0.61, −0.38 |
| Compound | 12·2DMF | 14 | 19 |
|---|---|---|---|
| CCDC number | 2245486 | 2245487 | 2245488 |
| Formula | C36H38N2O6 | C48H40O8 | C33H29NO5Zn |
| Mr | 594.68 | 392.43 | 584.94 |
| Crystal size (mm) | 0.2 × 0.15 × 0.1 | 0.2 × 0.15 × 0.1 | 0.08 × 0.04 × 0.005 |
| Crystal system | monoclinic | monoclinic | monoclinic |
| Space group | P21/c | C2/c | I2/m |
| Temperature (°C) | −173 | −173 | −173 |
| a (Å) | 17.3701(4) | 35.9642(3) | 10.44383(15) |
| b (Å) | 6.96876(14) | 13.38270(12) | 22.4818(3) |
| c (Å) | 13.2806(3) | 7.62745(7) | 11.21302(17) |
| α (°) | 90 | 90 | 90 |
| β (°) | 108.452(2) | 93.7660(8) | 99.6841(14) |
| γ (°) | 90 | 90 | 90 |
| V (Å3) | 1524.95 | 3663.15 | 2595.26 |
| Z | 2 | 4 | 4 |
| Dx (Mg m−3) | 1.295 | 1.350 | 1.497 |
| λ (Å) | 0.71073 (Mo Kα) | 1.54184 (Cu Kα) | 1.54184 (Cu Kα) |
| μ (mm−1) | 0.09 | 0.74 | 1.7 |
| Transmissions | 0.780–1.000 | 0.709–1.000 | 0.752–1.000 |
| F(000) | 632 | 1568 | 1216 |
| 2θmax | 80.5 | 155.2 | 155.0 |
| Refl. measured | 149,470 | 89,000 | 62,900 |
| Refl. indep. | 9604 | 3893 | 2834 |
| Rint | 0.047 | 0.054 | 0.033 |
| Parameters | 230 | 292 | 203 |
| Restraints | 19 | 24 | 3 |
| wR(F2, all refl.) | 0.116 | 0.116 | 0.069 |
| R(F, >4σ(F)) | 0.037 | 0.051 | 0.028 |
| S | 1.04 | 1.18 | 1.06 |
| Max. Δp (e Å−3) | 0.61, −0.29 | 0.31, −0.25 | 0.34, −0.52 |
| Zn(1)-O(3) | 1.9879(14) |
| Zn(1)-O(2)#1 | 2.0356(10) |
| Zn(1)-O(2) | 2.0356(10) |
| Zn(1)-O(1)#2 | 2.0499(11) |
| Zn(1)-O(1)#3 | 2.0499(11) |
| Zn(1)···Zn(1)#3 | 2.9182(5) |
| O(3)-Zn(1)-O(2)#1 | 98.89(4) |
| O(3)-Zn(1)-O(2) | 98.89(4) |
| O(2)#1-Zn(1)-O(2) | 85.64(6) |
| O(3)-Zn(1)-O(1)#2 | 100.21(5) |
| O(2)#1-Zn(1)-O(1)#2 | 92.66(5) |
| O(2)-Zn(1)-O(1)#2 | 160.85(5) |
| O(3)-Zn(1)-O(1)#3 | 100.21(5) |
| O(2)#1-Zn(1)-O(1)#3 | 160.85(5) |
| O(2)-Zn(1)-O(1)#3 | 92.67(5) |
| O(1)#2-Zn(1)-O(1)#3 | 82.71(7) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Birsa, M.L.; Hopf, H.; Jones, P.G.; Sarbu, L.G.; Bahrin, L.G. [2.2]Paracyclophane Derivatives as Building Blocks for Coordination Polymers. Materials 2023, 16, 4051. https://doi.org/10.3390/ma16114051
Birsa ML, Hopf H, Jones PG, Sarbu LG, Bahrin LG. [2.2]Paracyclophane Derivatives as Building Blocks for Coordination Polymers. Materials. 2023; 16(11):4051. https://doi.org/10.3390/ma16114051
Chicago/Turabian StyleBirsa, Mihail Lucian, Henning Hopf, Peter G. Jones, Laura Gabriela Sarbu, and Lucian Gabriel Bahrin. 2023. "[2.2]Paracyclophane Derivatives as Building Blocks for Coordination Polymers" Materials 16, no. 11: 4051. https://doi.org/10.3390/ma16114051