Simulation of Internal Defects in TKX-50 Crystals

Abstract
:1. Introduction
2. Calculation Models and Methods
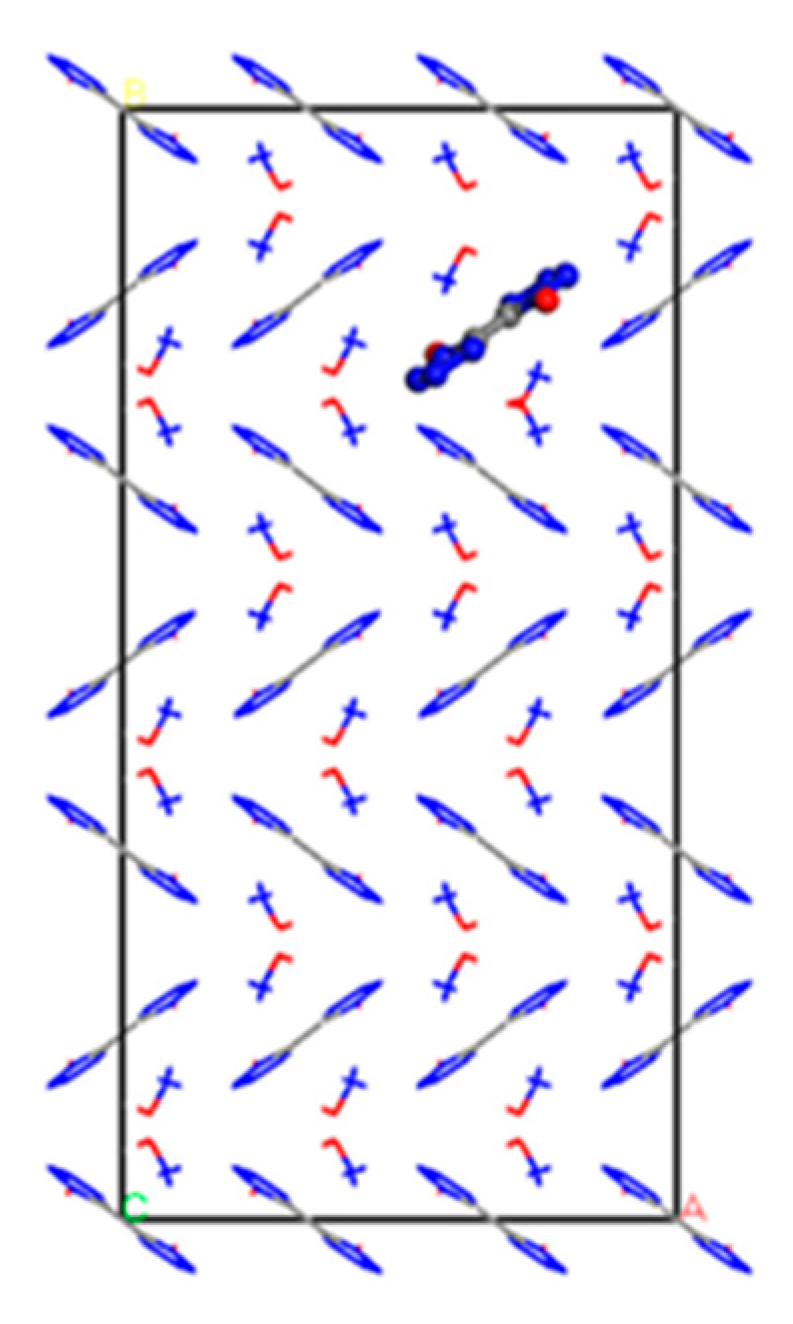
2.1. Establishment of the Initial Model of TKX-50 Crystal
2.2. Modeling of TKX-50 Crystal Defects
2.3. Calculation Conditions
3. Analysis of Results
3.1. Equilibrium Discriminant and Equilibrium Structure
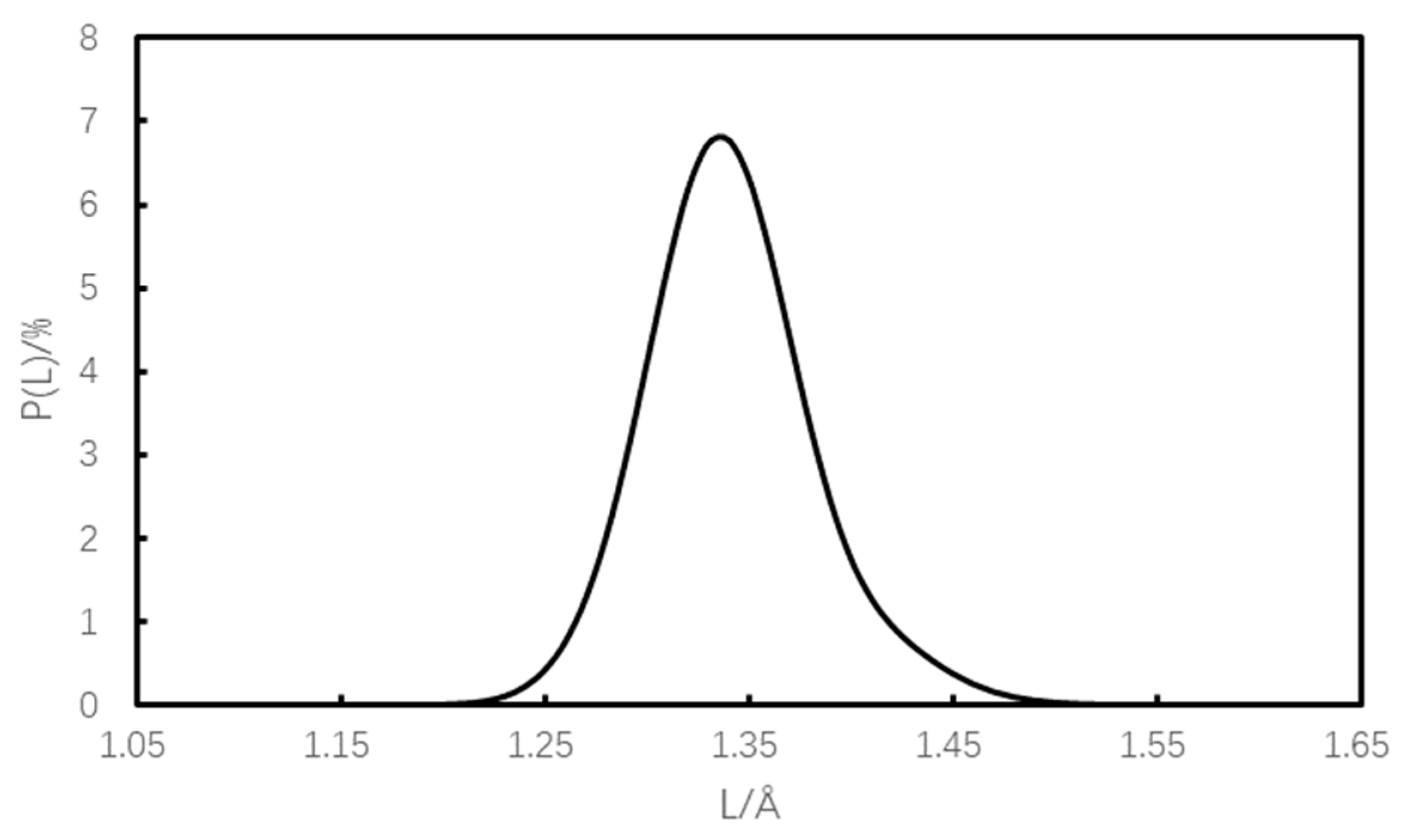
3.2. Lead Key Build Length
3.3. Bonded Diatomic Action Energy
3.4. Internal Cohesive Energy Density
3.5. Density
4. Conclusions
- The minimum bond length of defect-free crystals is 1.550 Å, the maximum bond-linked diatomic energy is 187.49 kcal·mol−1, and the maximum cohesive energy density is 0.764 kJ·cm−3; the bond length of defective crystals increases compared with that of defect-free crystals, with an increase of 1.42~36.77%; the bond-linked diatomic energy and cohesive energy density decrease, with decreases of 4.40~35.23% and 2.75~13.48%, respectively. The decrease in bonding diatomic interaction energy and cohesion energy density is 4.40~35.23% and 2.75~13.48%, respectively. This shows that with an increase in initiation bond length and a decrease in bond-linked diatomic energy and cohesive energy density, the sensitivity of the explosive increases and the safety decreases.
- The density of TKX-50 crystal without defect-type crystal is a maximum of 2.0590 g·cm−3, the activation bond ratio (Ract) is a minimum of 14.56%, and the density of defective crystal gradually decreases and shrinks by 3.70–9.67%; as the defect rate of crystal rises, the activation bond ratio (Ract) of the N-N bond, which is larger than the average bond length, also rises, and the crystal density decreases, which indicates that the density of TKX-50 crystals is related to the defect rate, and the larger the defect rate, the lower the density, indicating the increased sensitivity and weakened safety of the explosive.
- In the defect model without solvent doping, vacancy defects are more likely to affect the explosive susceptibility compared with dislocation defects, thus reducing the safety of explosives; among the seven solvent-doped defect models, N-methylpyrrolidone (NMP) doped defects have the largest bond length of 2.120 Å, the smallest bond-linked diatomic action energy, and cohesion energy density and density, which are 121 kcal·mol−1, 0.630 kJ·cm−3, and 1.8688 g·cm−3, so it has the most significant effect on the explosive sensitivities and is the most likely to reduce the safety of explosives.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ma, F. Study and Preparation of Spherification of TKX-50; North University of China: Taiyuan, China, 2019. [Google Scholar]
- Cao, R. Study on Crystallization Process of TKX-50 Spherification; North University of China: Taiyuan, China, 2021. [Google Scholar]
- Li, T. Research on Control Technology of Crystallization Spherification of TKX-50; North University of China: Taiyuan, China, 2022. [Google Scholar]
- Mi, X. Crystallization of a New High Energy Explosive TKX-50; North University of China: Taiyuan, China, 2015. [Google Scholar]
- Yu, E. Molecular Dynamics Simulations for 5,5′-bistetrazole-1,1′-diolate (TKX-50) and its PBXs. R. Soc. Chem. 2013, 6, 20034–20041. [Google Scholar] [CrossRef]
- Li, J.; Jin, S.; Lan, G. Reactive molecular dynamics simulations on the thermal decompositions and oxidations of TKX-50 and twinned TKX-50. Crystengcomm 2020, 50, 2593–2600. [Google Scholar] [CrossRef]
- Xiong, S.; Chen, S.; Jin, S. Molecular dynamic simulations on TKX-50/HMX cocrystal. RSC Adv. 2017, 7, 6795–6799. [Google Scholar] [CrossRef]
- Hang, G.; Yu, W.; Wang, T. Theoretical study on the effect of crystal defects on the properties of CL-20/NQ eutectic explosive. Energ. Mater. 2018, 10, 835–842. [Google Scholar]
- Yan, P.; Zhao, X.; Rui, J. Molecular Dynamics Simulation of The Influence of RDX Internal Defects On Sensitivity. Crystals 2021, 11, 329. [Google Scholar] [CrossRef]
- Vidal-Limon, A.; Toala, J.E.A.; Liceaga, A.M. Integration of Molecular Docking Analysis and Molecular Dynamics Simulations for Studying Food Proteins and Bioactive Peptides. J. Agric. Food Chem. 2022, 70, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Zarenezhad, E.; Behmard, E.; Rad, M.N.S. Synthesis, Cytotoxic Evaluation, Molecular Docking Studies and Molecular Dynamic Simulation of some Metronidazole Analogues. J. Mol. Struct. 2023, 1284, 135378. [Google Scholar] [CrossRef]
- Caliskan, M.; Mandaci, S.Y.; Uversky, V.N.; Coskuner-Weber, O. Secondary Structure Dependence of Amyloid-β(1–40) On Simulation Techniques and Force Field Parameters. Chem. Biol. Drug Des. 2021, 97, 1100–1108. [Google Scholar] [CrossRef]
- Zhang, D.; Li, Y.; Zhang, J. Influence of Conversion On Dielectric Constant of Dicyandiamide Cured Epoxy Resin: A Molecular Dynamic Simulation and Experiment Study. Polymer 2023, 267, 125645. [Google Scholar] [CrossRef]
- Ragab, S.S.; Abdelraof, M.; Sweed, A.M.K. Design, Synthesis, Molecular Dynamic Simulation Studies, and Antibacterial Evaluation of New Spirocyclic Aminopyrimidines. J. Mol. Struct. 2023, 1278, 134912. [Google Scholar] [CrossRef]
- Guo, Y.; Chen, X.; Cao, X. Prediction and Validation of Monoclonal Antibodies Separation in Aqueous Two-phase System Using Molecular Dynamic Simulation. J. Chromatogr. A 2023, 1694, 463921. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; He, Y.; Zhai, M. Molecular Dynamic Simulations of Methane Hydrate Formation Between Solid Surfaces: Implications for Methane Storage. Energy 2023, 262, 125511. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Y.; Wang, B. Solubility, Dissolution Properties and Molecular Dynamic Simulation of 2,6-bis(picrylamino)-3,5-dinitropyridine in Pure and Binary Solvents. J. Mol. Liq. 2022, 368, 120567. [Google Scholar] [CrossRef]
- Ganji, M.D.; Fereidoon, A.; Ahangari, M.G. Elastic Properties of Swcnts with Curved Morphology: Density Functional Tight Binding Based Treatment. Solid State Commun. 2012, 152, 1526–1530. [Google Scholar] [CrossRef]
- Shreider, Y.A. (Ed.) Chapter I-Principles of The Monte Carlo Method. In The Monte Carlo Method; Pergamon: Oxford, UK, 1966; pp. 1–90. [Google Scholar]
- Jin, H.; Andritsch, T.; Tsekmes, I.A.; Kochetov, R.; Morshuis, P.H.F.; Smit, J.J. Properties of mineral oil based silica nanofluids. IEEE Trans. Dielectr. Electr. Insul. 2014, 21, 1100–1108. [Google Scholar]
- Mashhadzadeh, A.H.; Ahangari, M.G.; Dadrasi, A.; Fathalian, M. Theoretical studies on the mechanical and electronic properties of 2D and 3D structures of Beryllium-Oxide graphene and graphene nanobud. Appl. Surf. Sci. 2019, 476, 36–48. [Google Scholar] [CrossRef]
- Liu, H.; Liu, N.; Wang, D.; Ren, J. Ab Initio Molecular Dynamics Study of Adsorption of Hydroxyl Groups On Graphene Surface. Chin. J. Chem. Phys. 2021, 34, 777–784. [Google Scholar] [CrossRef]
- Srivastava, N.; Majumder, C. Novel Biofiltration Methods for the Treatment of Heavy Metals from Industrial Wastewater. J. Hazard. Mater. 2008, 151, 1–8. [Google Scholar] [CrossRef]
- Huo, E.; Zhang, S.; Bai, M. Pyrolysis Mechanism Study of N-heptane as an Endothermic Hydrocarbon Fuel: A Reactive Molecular Dynamic Simulation and Density Functional Theory Calculation Study. Comput. Theor. Chem. 2022, 1211, 113696. [Google Scholar] [CrossRef]
- Feng, X.; Li, F.; Shi, T. Molecular Dynamic Simulation: Conformational Properties of Single-stranded Curdlan in Aqueous Solution. Carbohydr. Polym. 2020, 250, 116906. [Google Scholar] [CrossRef]
- Ebrahimi, S.; Meunier, M.; Soldera, A. Molecular Dynamics Simulation of the Dynamical Mechanical Analysis of Polybutadiene. Polym. Test. 2022, 111, 107585. [Google Scholar] [CrossRef]
- Chen, C.; Zhong, C.; Zu, Q. Molecular Dynamic Simulations Study On the Structure and Properties of Li2O-containing Magnesium Aluminosilicate Glasses. Mater. Commun. 2022, 32, 103945. [Google Scholar] [CrossRef]
- Sharif, M.; Zhang, T.; Zhang, Z. Evaluation of CO2 Absorption Performance by Molecular Dynamic Simulation for Mixed Secondary and Tertiary Amines. Int. J. Greenh. Gas Control 2020, 97, 103059. [Google Scholar] [CrossRef]
- Zheng, R.; Wang, Z.; Negahban, S. Structural and Dynamic Analyses of CH4-C2H6-CO2 Hydrates Using Thermodynamic Modeling and Molecular Dynamic Simulation. J. Chem. Thermodyn. 2022, 169, 106749. [Google Scholar] [CrossRef]
- Mi, S.; Fahs, A.; Bousseksou, A. Molecular Mechanics Simulations of Lattice Dynamical Properties of the Spin Crossover Complex[Fe(pyrazine)][Ni(CN)4]. Chem. Phys. Lett. 2022, 811, 140232. [Google Scholar] [CrossRef]
- Zhao, Q.; Gao, H.; Ouyang, D. Experimental Characterization and Molecular Dynamic Simulation of Ketoprofen-cyclodextrin Complexes. Chem. Phys. Lett. 2019, 736, 136802. [Google Scholar] [CrossRef]
- Hua, C.; Huang, M.; Huang, H. Characterization of internal defects and sensitivity of shock wave in RDX/HMX explosive crystals. Energ. Mater. 2010, 2, 152–156. [Google Scholar]
- Erofeev, L.N.; Tarasov, Y.P.; Kalmykov, Y.B.; Shu, Y.; Dubikhin, V.V.; Nazin, G.M. Crystal defects and stability of RDX. Russ. Chem. Bull. 2001, 50, 1000–1002. [Google Scholar] [CrossRef]
- Bouma, R.; Duvalois, W.; Heijden, A. Microscopic characterization of defect structure in RDX crystals. J. Microsc. 2013, 252, 263–274. [Google Scholar] [CrossRef]
- Kim, K.; Kim, K.-J. Quantitative Study on Crystal Defects Using the Relationship between Crystallization Parameters and Thermal Analysis. Cryst. Growth Des. 2018, 18, 5021–5028. [Google Scholar] [CrossRef]
- Ren, X.; Zhang, G. Calculation and Control of Crystal Form of 1,1′-Dihydroxy-5,5′—Tetrazolium Dihydroxyamine Salt. J. Explos. 2016, 39, 68–71. [Google Scholar]
- Zhou, T.; Chen, F.; Li, J.; Cao, D.; Wang, J. Molecular dynamics simulation of the growth morphology of TKX-50 in formic acid/water mixed solvents. Energetic Mater. 2020, 28, 865–873. [Google Scholar]
- Liu, Y.; Bi, F. Prediction of crystal morphology of 5,5′-ditetrazol-1,1′—Dihydroxyammonium under different growth conditions. Energ. Mater. 2018, 26, 210–217. [Google Scholar]
- Zhu, Z.; Jiang, Z.; Wang, P.; Lu, M. Synthesis and properties of 5,5′-ditetrazol-1,1′—Dioxodihydroxyammonium. Energ. Mater. 2014, 22, 332–336. [Google Scholar]
- Li, D. Spheroidization of 1,1′-Dihydroxy-5,5′—Tetrazolium Dihydroxyamine Salt; Central North University: Taiyuan, China, 2019. [Google Scholar]
- Bi, F.; Xiao, C. Synthesis and properties of 1,1′-dihydroxy-5,5′—Tetrazolium dihydroxyamine salt. Energ. Mater. 2014, 22, 272–273. [Google Scholar]
- Chen, F.; Zhou, T.; Li, J.; Wang, X.; Cao, D.; Wang, J.; Yang, Z. Crystal Morphology of Dihydroxylammonium 5,5′-bistetrazole-1,1′-diolate (tkx-50) Under Solvents System with Different Polarity Using Molecular Dynamics. Comput. Mater. Sci. 2019, 168, 48–57. [Google Scholar] [CrossRef]
- Mi, D.; Zhang, R. Preparation and Performance Characterization of Spheroidized TKX-50. J. Pyrotech. Explos. 2020, 15, 64–68. [Google Scholar]
- Ma, S.; Li, Y.; Li, Y.; Luo, Y. Research on structures, mechanical properties, and mechanical responses of TKX-50 and TKX-50 based PBX with molecular dynamics. J. Mol. Model. 2016, 22, 43. [Google Scholar] [CrossRef]
- Yu, C.; Yang, L.; Chen, H.; Qin, Y.; Wang, T.; Sun, W.; Wang, C. Microscale investigations of mechanical responses of TKX-50 based polymer bonded explosives using MD simulations. Comput. Mater. Sci. 2020, 172, 109287. [Google Scholar] [CrossRef]
- Ding, Q.; Jia, C.; Tang, H. Unified Non-fitting Formulation Representation of Thermodynamic Properties for Diatomic Substances. J. Mol. Liq. 2022, 371, 121088. [Google Scholar] [CrossRef]
- Almasi, M. Quantifying Intermolecular Interactions Between 1-hexyl-3-methylimidazolium Nitrate and 1-alkanol: Internal Pressure and Cohesive Energy Density Approach. Chem. Phys. 2020, 539, 110936. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, Y.; Lu, T. High Density Nano-grained Gd2zr2o7 Ceramic Prepared by Combined Cold and Microwave Sintering. Ceram. Int. 2022, 48, 26387–26392. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Lprob/Å | Lave/Å | Lmax/Å |
|---|---|---|---|
| 1 | 1.399 | 1.200 | 1.550 |
| 2 | 1.399 | 1.205 | 1.572 |
| 3 | 1.399 | 1.215 | 1.574 |
| 4 | 1.340 | 1.210 | 1.578 |
| 5 | 1.340 | 1.215 | 1.583 |
| 6 | 1.341 | 1.205 | 1.560 |
| 7 | 1.340 | 1.210 | 1.570 |
| 8 | 1.341 | 1.240 | 1.650 |
| 9 | 1.341 | 1.241 | 1.690 |
| 10 | 1.340 | 1.240 | 1.660 |
| 11 | 1.342 | 1.245 | 2.120 |
| Model | CED/kJ·cm−3 | vdW/kJ·cm−3 | Electrostatic/kJ·cm−3 |
|---|---|---|---|
| 1 | 0.764 | 0.059 | 0.705 |
| 2 | 0.738 | 0.055 | 0.683 |
| 3 | 0.736 | 0.054 | 0.682 |
| 4 | 0.733 | 0.052 | 0.681 |
| 5 | 0.683 | 0.045 | 0.638 |
| 6 | 0.743 | 0.055 | 0.688 |
| 7 | 0.729 | 0.056 | 0.673 |
| 8 | 0.718 | 0.055 | 0.663 |
| 9 | 0.714 | 0.054 | 0.660 |
| 10 | 0.711 | 0.053 | 0.658 |
| 11 | 0.661 | 0.031 | 0.630 |
| Model | ρ/g·cm−3 | Ract/% |
|---|---|---|
| 1 | 2.0590 | 14.56 |
| 2 | 1.9828 | 17.01 |
| 3 | 1.9446 | 20.72 |
| 4 | 1.9065 | 24.32 |
| 5 | 1.8598 | 29.49 |
| 6 | 1.9537 | 16.95 |
| 7 | 1.9410 | 20.13 |
| 8 | 1.9177 | 23.42 |
| 9 | 1.8960 | 25.02 |
| 10 | 1.9148 | 23.49 |
| 11 | 1.8688 | 15.74 |
| 12 | 1.8835 | 26.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, S.; Zhao, X.; Li, Y.; Ding, W.; Huang, J. Simulation of Internal Defects in TKX-50 Crystals. Materials 2023, 16, 4063. https://doi.org/10.3390/ma16114063
Qiu S, Zhao X, Li Y, Ding W, Huang J. Simulation of Internal Defects in TKX-50 Crystals. Materials. 2023; 16(11):4063. https://doi.org/10.3390/ma16114063
Chicago/Turabian StyleQiu, Siqi, Xue Zhao, Yuanyuan Li, Wenyuan Ding, and Junrui Huang. 2023. "Simulation of Internal Defects in TKX-50 Crystals" Materials 16, no. 11: 4063. https://doi.org/10.3390/ma16114063
APA StyleQiu, S., Zhao, X., Li, Y., Ding, W., & Huang, J. (2023). Simulation of Internal Defects in TKX-50 Crystals. Materials, 16(11), 4063. https://doi.org/10.3390/ma16114063