
Structural Evolution in Glassy Carbon Investigated Based on the Temperature Dependence of Young’s Modulus

Abstract
:1. Introduction
2. Experimental Methods
2.1. GC Fabricating Process
2.2. Characterizations
2.2.1. Thermogravimetric Analysis (TGA) and Thermomechanical Analysis (TMA)
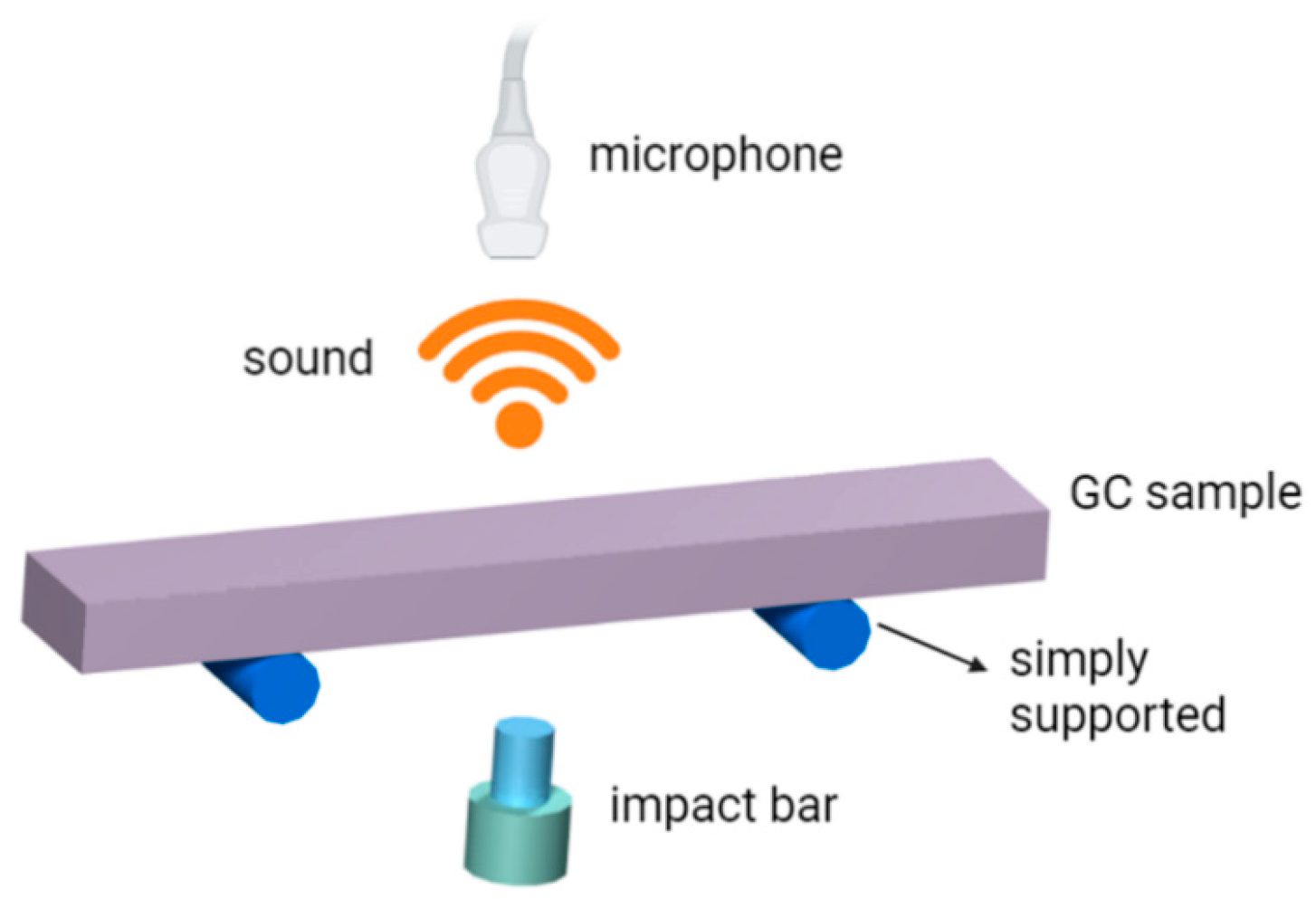
2.2.2. Impulse Excitation Technique (IET)
2.2.3. X-ray Diffraction (XRD) and Raman Spectroscopy
3. Results and Discussion
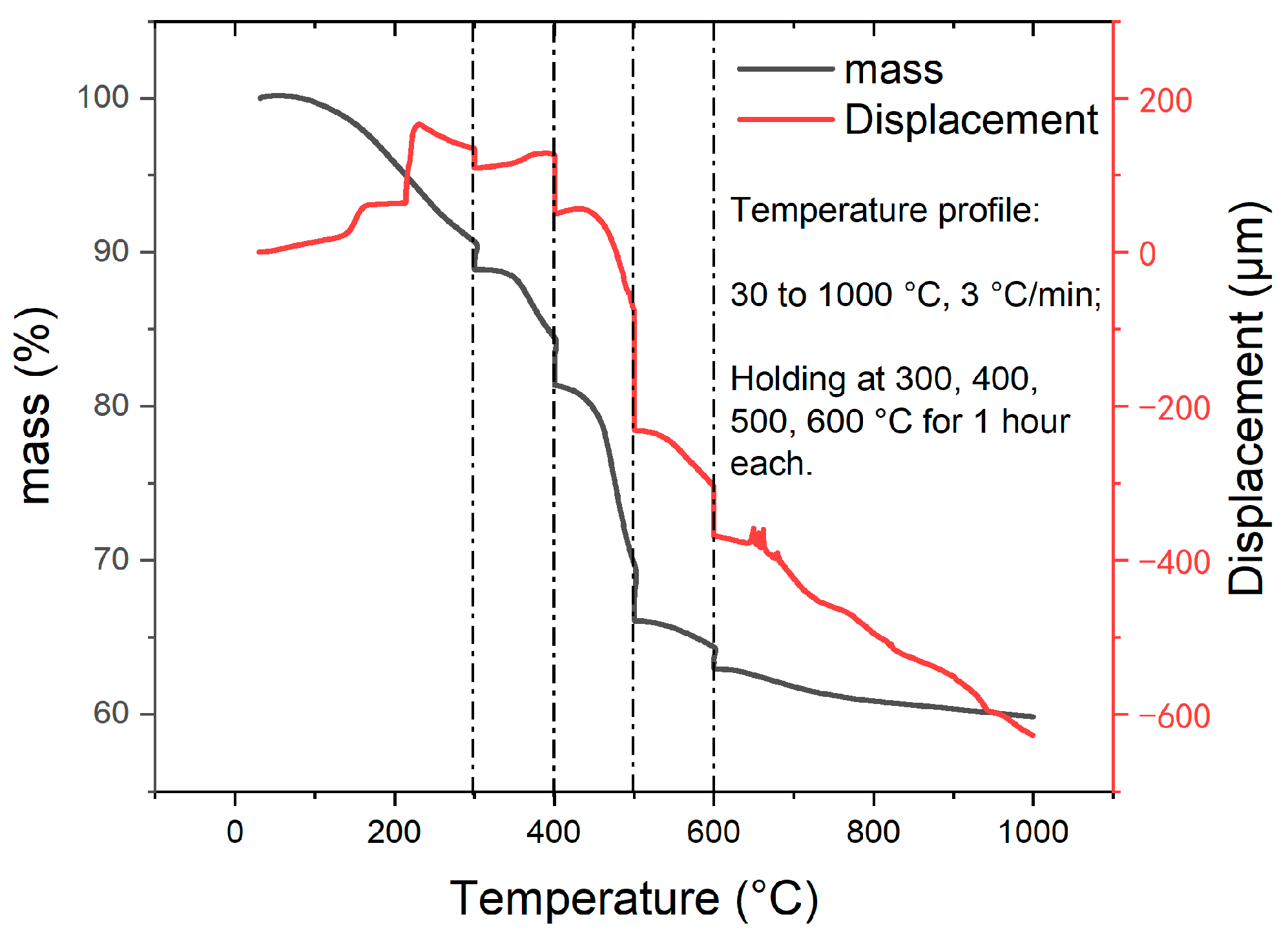
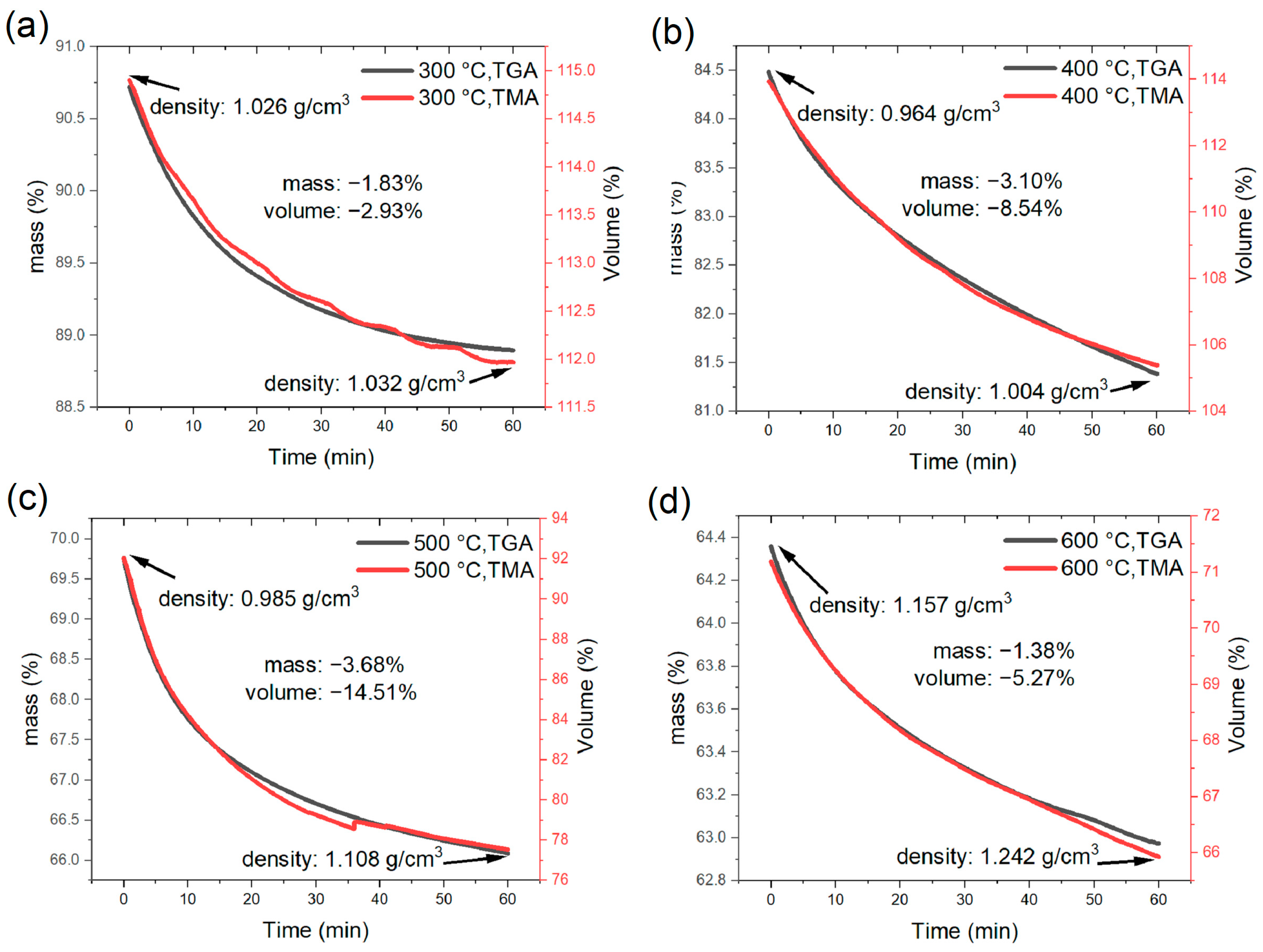
3.1. TMA and TGA Results of Pyrolysis
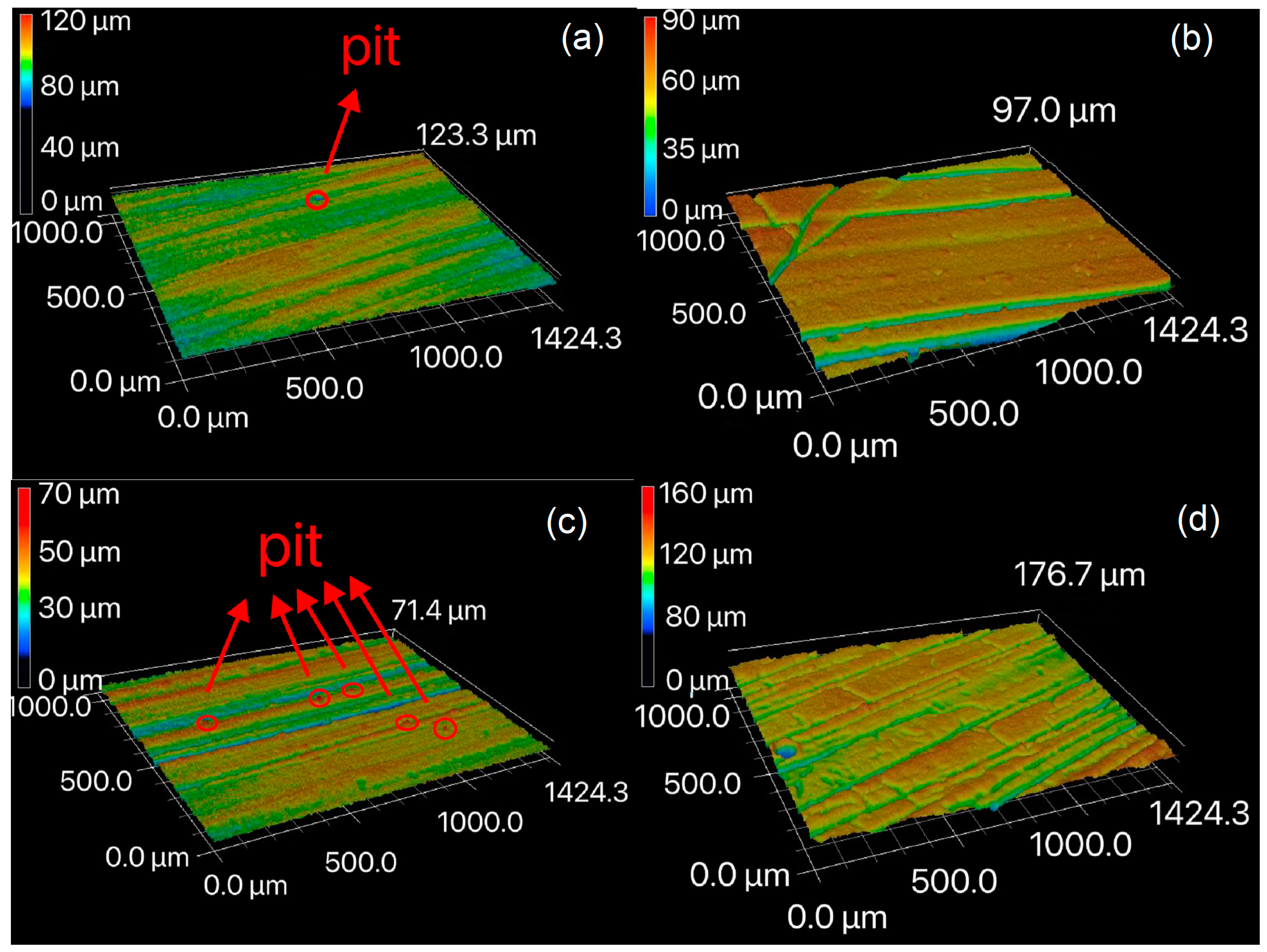
3.2. Surface Morphology Evolution after Pyrolysis
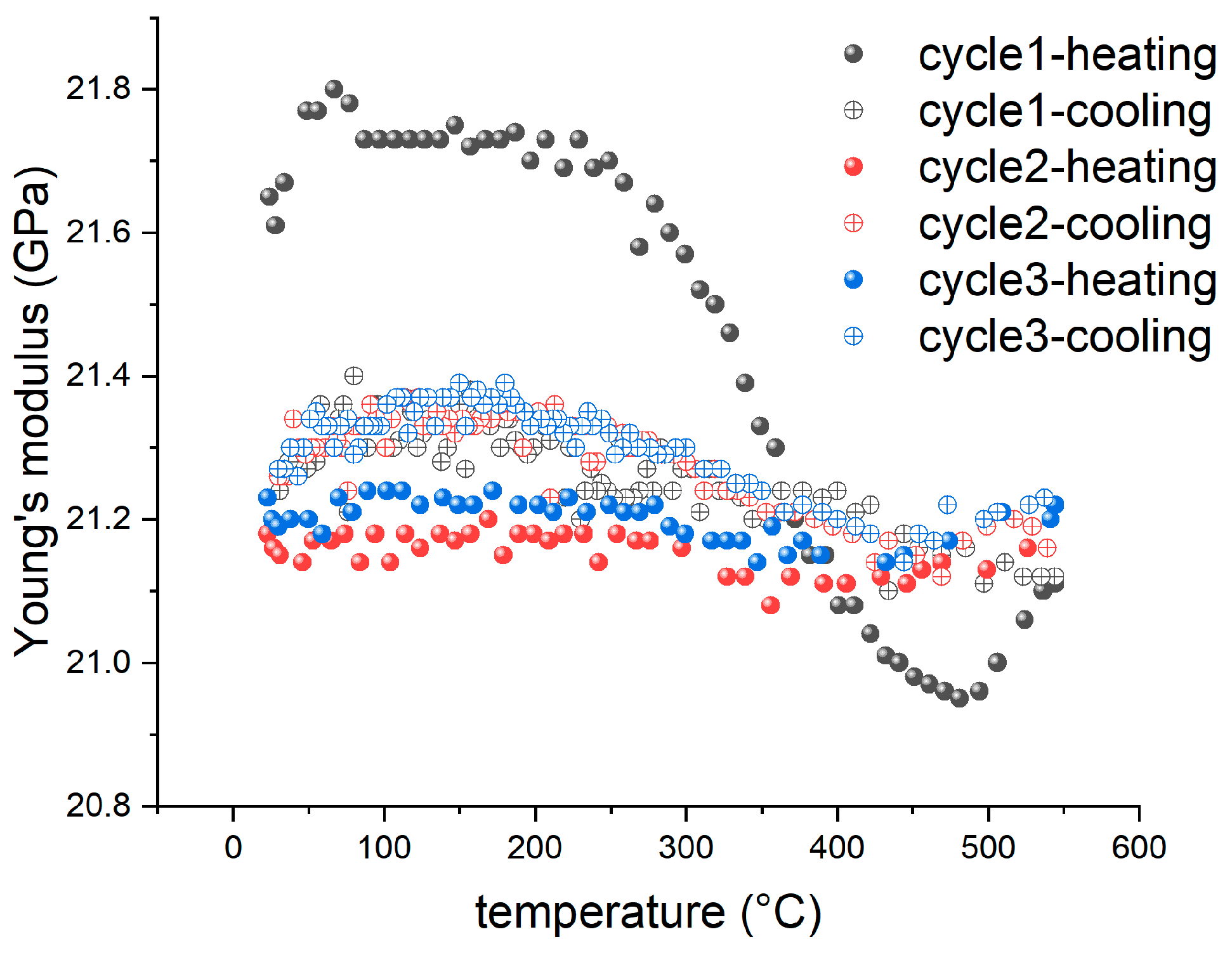
3.3. Comparison between IET and TGA
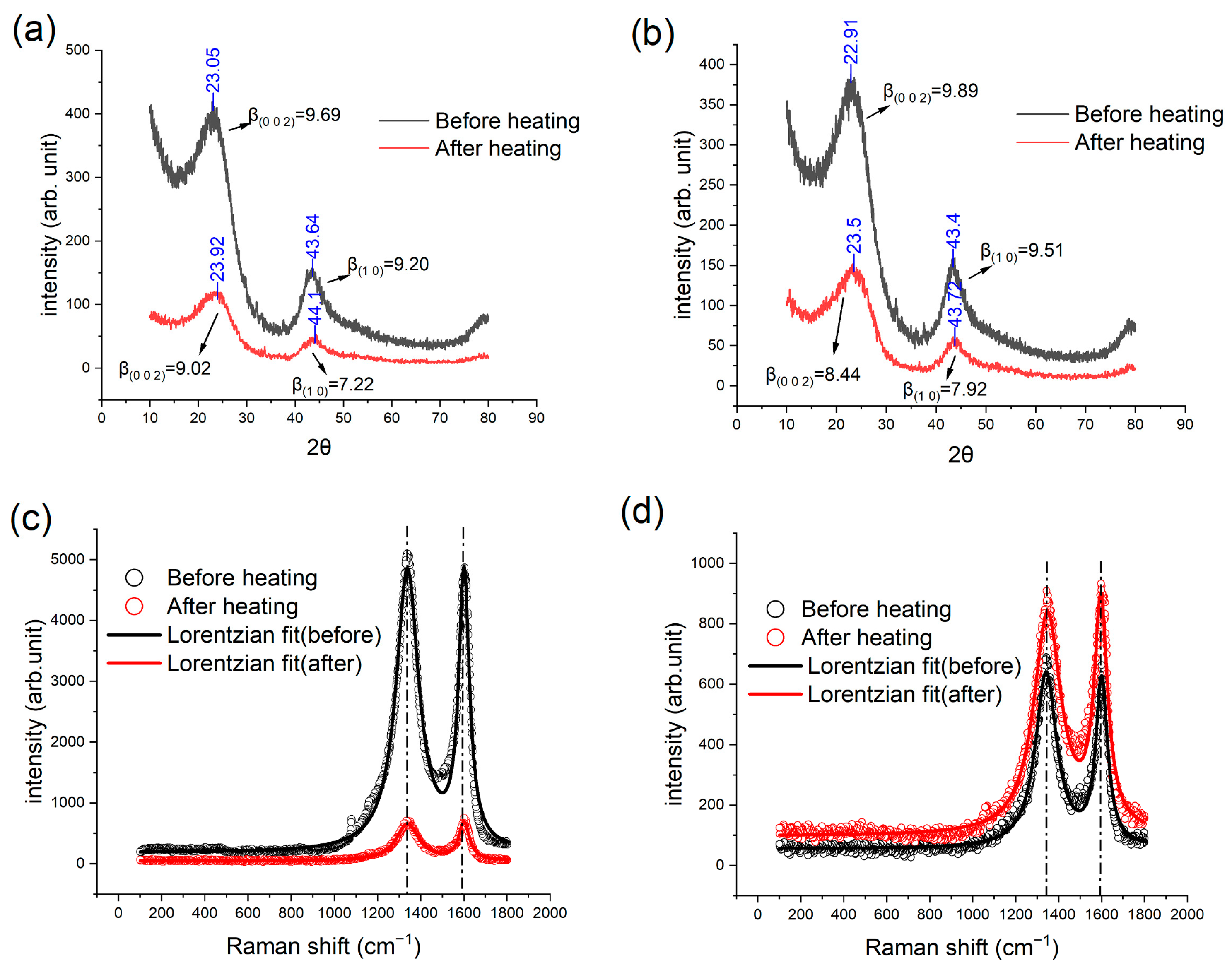
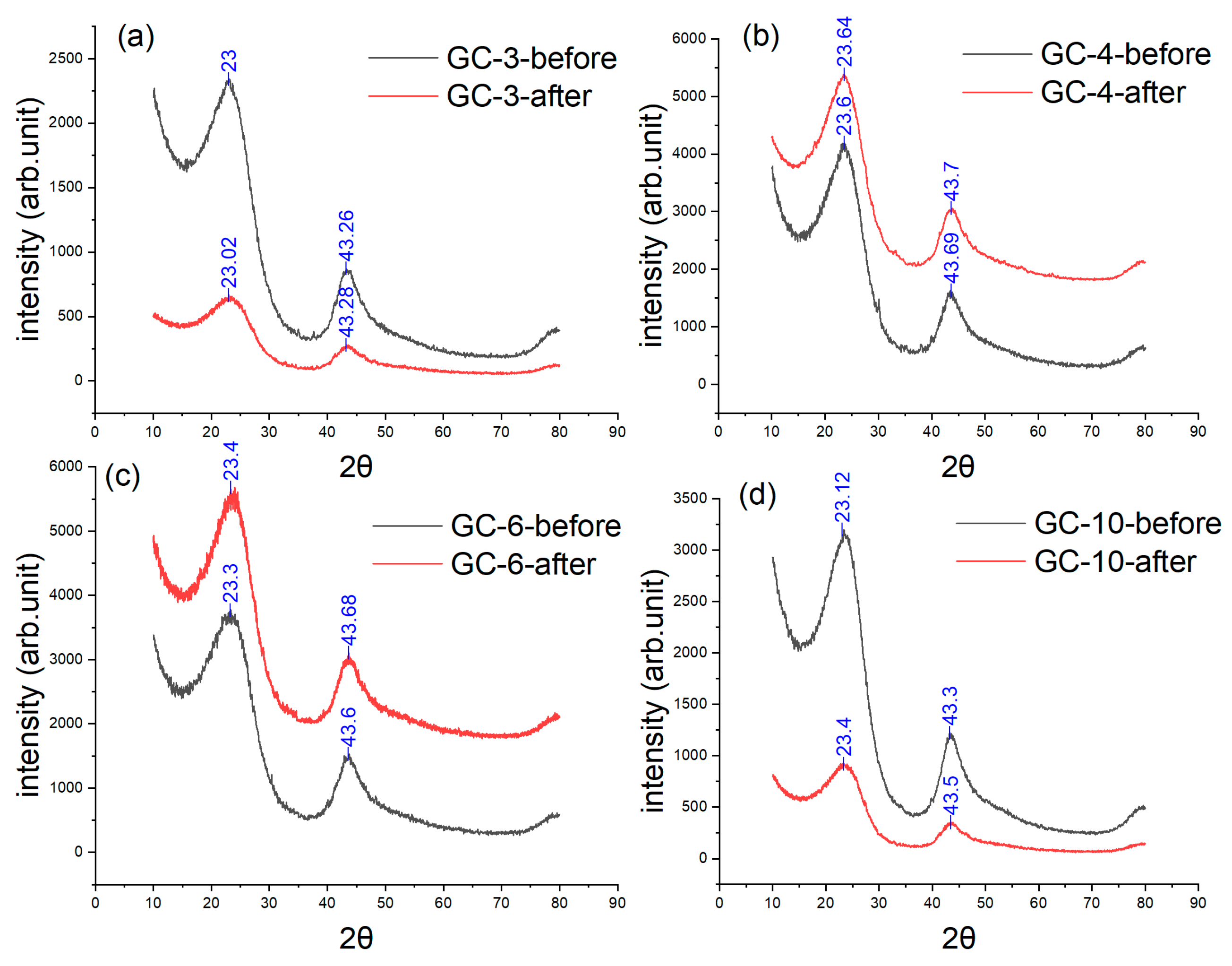
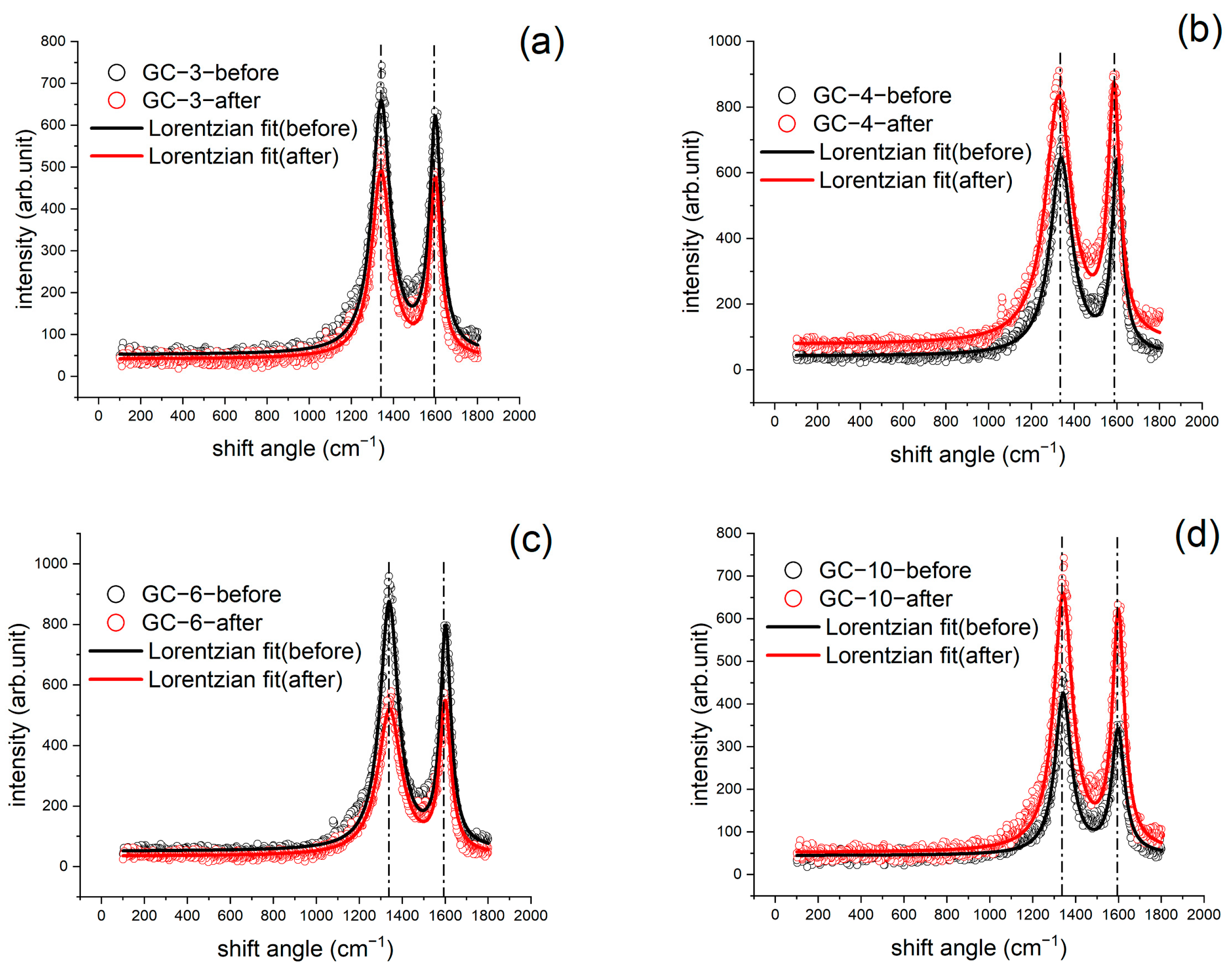
3.4. XRD and Raman Results
3.5. The Effect of Heating Rate
3.6. Thermal Enhancement
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, S.; Shi, T.; Zhan, X.; Xi, S.; Long, H.; Gong, B.; Li, J.; Cheng, S.; Huang, Y.; Tang, Z. Scalable fabrication of carbon-based MEMS/NEMS and their applications: A review. J. Micromech. Microeng. 2015, 25, 113001. [Google Scholar] [CrossRef]
- VanDersarl, J.J.; Mercanzini, A.; Renaud, P. Integration of 2D and 3D Thin Film Glassy Carbon Electrode Arrays for Electrochemical Dopamine Sensing in Flexible Neuroelectronic Implants. Adv. Funct. Mater. 2015, 25, 78–84. [Google Scholar] [CrossRef]
- Mukhopadhyay, K.; Sharon, M. Glassy carbon from camphor—A natural source. Mater. Chem. Phys. 1997, 49, 105–109. [Google Scholar] [CrossRef]
- Botelho, E.C.; Scherbakoff, N.; Rezende, M.C. Porosity control in glassy carbon by rheological study of the furfuryl resin. Carbon 2001, 39, 45–52. [Google Scholar] [CrossRef]
- Lewis, J.; Redfern, B.; Cowlard, F. Vitreous carbon as a crucible material for semiconductors. Solid-State Electron. 1963, 6, 251–254. [Google Scholar] [CrossRef]
- Noda, T.; Inagaki, M. The structure of glassy carbon. Bull. Chem. Soc. Jpn. 1964, 37, 1534–1538. [Google Scholar] [CrossRef]
- Youn, S.; Takahashi, M.; Goto, H.; Maeda, R. Fabrication of micro-mold for glass embossing using focused ion beam, femto-second laser, eximer laser and dicing techniques. J. Mater. Process. Technol. 2007, 187, 326–330. [Google Scholar] [CrossRef]
- Ju, J.; Han, Y.; Seok, J.; Kim, S.-M. Development of low-cost and large-area nanopatterned vitreous carbon stamp for glass nanoreplication. In Proceedings of the 2012 12th IEEE International Conference on Nanotechnology (IEEE-NANO), Birmingham, UK, 20–23 August 2012. [Google Scholar]
- Takahashi, M.; Sugimoto, K.; Maeda, R. Nanoimprint of Glass Materials with Glassy Carbon Molds Fabricated by Focused-Ion-Beam Etching. Jpn. J. Appl. Phys. 2005, 44, 5600. [Google Scholar] [CrossRef]
- Musameh, M.; Wang, J.; Merkoci, A.; Lin, Y. Low-potential stable NADH detection at carbon-nanotube-modified glassy carbon electrodes. Electrochem. Commun. 2002, 4, 743–746. [Google Scholar] [CrossRef]
- Murugan, P.; Nagarajan, R.D.; Sundramoorthy, A.K.; Ganapathy, D.; Atchudan, R.; Nallaswamy, D.; Khosla, A. Electrochemical Detection of H2O2 Using an Activated Glassy Carbon Electrode. ECS Sens. Plus 2022, 1, 034401. [Google Scholar] [CrossRef]
- Hucke, E.E.; Fuys, R.A.; Craig, R.G. Glassy carbon: A potential dental implant material. J. Biomed. Mater. Res. 1973, 7, 263–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S. Glassy Carbon: A Promising Material for Micro- and Nanomanufacturing. Materials 2018, 11, 1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, G.M.; Kawamura, K. Polymeric Carbons: Carbon Fibre, Glass and Char; Cambridge University Press: Cambridge, UK, 1976. [Google Scholar]
- Cowlard, F.; Lewis, J. Vitreous carbon—A new form of carbon. J. Mater. Sci. 1967, 2, 507–512. [Google Scholar] [CrossRef]
- Yamada, S.; Sato, H.; Ishii, T. Eigenschaften und Verwendung von glasartigem Kohlenstoff. Carbon 1964, 2, 253–260. [Google Scholar] [CrossRef]
- Liu, W.; Liu, M.; Zhang, L. Oxidation-induced mechanical deterioration and hierarchical cracks in glassy carbon. Carbon 2016, 100, 178–186. [Google Scholar] [CrossRef]
- Nakamizo, M.; Kammereck, R.; Walker, P. Laser raman studies on carbons. Carbon 1974, 12, 259–267. [Google Scholar] [CrossRef]
- Trick, K.A.; Saliba, T.E. Mechanisms of the pyrolysis of phenolic resin in a carbon/phenolic composite. Carbon 1995, 33, 1509–1515. [Google Scholar] [CrossRef]
- Jiang, H.; Wang, J.; Wu, S.; Yuan, Z.; Hu, Z.; Wu, R.; Liu, Q. The pyrolysis mechanism of phenol formaldehyde resin. Polym. Degrad. Stab. 2012, 97, 1527–1533. [Google Scholar] [CrossRef]
- Lee, M.R. Transmission electron microscopy (TEM) of Earth and planetary materials: A review. Miner. Mag. 2010, 74, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, M.; Sigesato, G. A review of focused ion beam technology and its applications in transmission electron microscopy. Microscopy 2004, 53, 527–536. [Google Scholar] [CrossRef]
- Castagnola, E.; Vahidi, N.W.; Nimbalkar, S.; Rudraraju, S.; Thielk, M.; Zucchini, E.; Cea, C.; Carli, S.; Gentner, T.Q.; Ricci, D.; et al. In Vivo Dopamine Detection and Single Unit Recordings Using Intracortical Glassy Carbon Microelectrode Arrays. MRS Adv. 2018, 3, 1629–1634. [Google Scholar] [CrossRef] [PubMed]
- Hassan, Y.M.; Caviglia, C.; Hemanth, S.; Mackenzie, D.M.A.; Petersen, D.H.; Keller, S.S. Pyrolytic carbon microelectrodes for impedance based cell sensing. ECS Trans. 2016, 72, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Madou, M. A new approach to gas sensing with nanotechnology. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2012, 370, 2448–2473. [Google Scholar] [CrossRef] [PubMed]
- Ghiasi, T.; Ahmadi, S.; Ahmadi, E.; Olyai, M.R.T.B.; Khodadadi, Z. Novel electrochemical sensor based on modified glassy carbon electrode with graphene quantum dots, chitosan and nickel molybdate nanocomposites for diazinon and optimal design by the Taguchi method. Microchem. J. 2020, 160, 105628. [Google Scholar] [CrossRef]
- Bauer, J.; Schroer, A.; Schwaiger, R.; Kraft, O. Approaching theoretical strength in glassy carbon nanolattices. Nat. Mater. 2016, 15, 438–443. [Google Scholar] [CrossRef]
- Hauert, A.; Rossoll, A.; Mortensen, A. Young’s modulus of ceramic particle reinforced aluminium: Measurement by the Impulse Excitation Technique and confrontation with analytical models. Compos. Part A: Appl. Sci. Manuf. 2009, 40, 524–529. [Google Scholar] [CrossRef]
- Kim, Y.K.; Haq, M.R.; Kim, S.-M. Glass molding of all glass Fresnel lens with vitreous carbon micromold. Opt. Express 2019, 27, 1553–1562. [Google Scholar] [CrossRef]
- Hancock, B.C.; Dupuis, Y.; Thibert, R. Determination of the viscosity of an amorphous drug using thermomechanical analysis (TMA). Pharm. Res. 1999, 16, 672–675. [Google Scholar] [CrossRef]
- Hu, H.; Wang, W.; Jiang, L.; Liu, L.; Zhang, Y.; Yang, Y.; Wang, J. Curing mechanism of resole phenolic resin based on variable temperature FTIR spectra and thermogravimetry-mass spectrometry. Polym. Polym. Compos. 2022, 30, 09673911221102114. [Google Scholar] [CrossRef]
- Fitzer, E.; Schäfer, W. The effect of crosslinking on the formation of glasslike carbons from thermosetting resins. Carbon 1970, 8, 353–364. [Google Scholar] [CrossRef]
- Krahn, J.; Morgenstern, N. The ultimate frictional resistance of rock discontinuities. Int. J. Rock Mech. Min. Sci. Geomech. Abstr. 1979, 16, 127–133. [Google Scholar] [CrossRef]
- Diss, P.; Lamon, J.; Carpentier, L.; Loubet, J.; Kapsa, P. Sharp indentation behavior of carbon/carbon composites and varieties of carbon. Carbon 2002, 40, 2567–2579. [Google Scholar] [CrossRef]
- Fielda, J.; Swain, M. The indentation characterisation of the mechanical properties of various carbon materials: Glassy carbon, coke and pyrolytic graphite. Carbon 1996, 34, 1357–1366. [Google Scholar] [CrossRef]
- Sakai, M.; Hanyu, H.; Inagaki, M. Indentation-Induced Contact Deformation and Damage of Glasslike Carbon. J. Am. Ceram. Soc. 1995, 78, 1006–1012. [Google Scholar] [CrossRef]
- Garion, C. Mechanical Properties for Reliability Analysis of Structures in Glassy Carbon. World J. Mech. 2014, 4, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Shiell, T.B.; Wong, S.; Yang, W.; Tanner, C.A.; Haberl, B.; Elliman, R.; McKenzie, D.; McCulloch, D.; Bradby, J.E. The composition, structure and properties of four different glassy carbons. J. Non-Cryst. Solids 2019, 522, 119561. [Google Scholar] [CrossRef]
- Soukup, L.; Gregora, I.; Jastrabik, L.; Koňáková, A. Raman spectra and electrical conductivity of glassy carbon. Mater. Sci. Eng. B 1992, 11, 355–357. [Google Scholar] [CrossRef]
- Eto, M.; Oku, T.; Konishi, T. High temperature young’s modulus of a fine-grained nuclear graphite oxidized or prestressed to various levels. Carbon 1991, 29, 11–21. [Google Scholar] [CrossRef]
- Monserrat, B.; Drummond, N.D.; Needs, R.J. Anharmonic vibrational properties in periodic systems: Energy, electron-phonon coupling, and stress. Phys. Rev. B 2013, 87, 144302. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Jiang, H.; Jiang, N. Study on the pyrolysis of phenol-formaldehyde (PF) resin and modified PF resin. Thermochim. Acta 2009, 496, 136–142. [Google Scholar] [CrossRef]
- Pawlyta, M.; Rouzaud, J.-N.; Duber, S. Raman microspectroscopy characterization of carbon blacks: Spectral analysis and structural information. Carbon 2015, 84, 479–490. [Google Scholar] [CrossRef]
- Tomita, S.; Burian, A.; Dore, J.C.; LeBolloch, D.; Fujii, M.; Hayashi, S. Diamond nanoparticles to carbon onions transformation: X-ray diffraction studies. Carbon 2002, 40, 1469–1474. [Google Scholar] [CrossRef]
- Surekha, G.; Krishnaiah, K.V.; Ravi, N.; Suvarna, R.P. FTIR, Raman and XRD analysis of graphene oxide films prepared by modified Hummers method. J. Phys. Conf. Ser. 2020, 1495, 012012. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 2000, 61, 14095–14107. [Google Scholar] [CrossRef] [Green Version]
- Tuinstra, F.; Koenig, J.L. Raman spectrum of graphite. J. Chem. Phys. 1970, 53, 1126–1130. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surface Roughness | Ra (μm) | Rq (μm) |
|---|---|---|
| 6 h-cured precursor | 4.1396 | 5.1740 |
| GC obtained from 6 h-cured precursor | 8.9007 | 12.8469 |
| 22 h-cured precursor | 4.4794 | 5.9644 |
| GC obtained from 22 h-cured precursor | 7.7515 | 9.6199 |
| Raman Results (ID/IG) | GC-3 | GC-4 | GC-6 | GC-10 |
|---|---|---|---|---|
| Before IET | 1.1726 | 1.1099 | 1.180 | 1.329 |
| After IET | 1.1436 | 1.0120 | 1.010 | 1.107 |
| Electrical Conductivity(S/cm) | GC-3 | GC-4 | GC-6 | GC-10 |
|---|---|---|---|---|
| Before IET | 99.133 | 105.51 | 108.502 | 110.313 |
| After IET | 53.345 | 40.324 | 35.336 | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Dang, Y.; Ruan, H. Structural Evolution in Glassy Carbon Investigated Based on the Temperature Dependence of Young’s Modulus. Materials 2023, 16, 4794. https://doi.org/10.3390/ma16134794
Yang Y, Dang Y, Ruan H. Structural Evolution in Glassy Carbon Investigated Based on the Temperature Dependence of Young’s Modulus. Materials. 2023; 16(13):4794. https://doi.org/10.3390/ma16134794
Chicago/Turabian StyleYang, Yi, Yanpei Dang, and Haihui Ruan. 2023. "Structural Evolution in Glassy Carbon Investigated Based on the Temperature Dependence of Young’s Modulus" Materials 16, no. 13: 4794. https://doi.org/10.3390/ma16134794