
Interplay of Oxidative Stress, Inflammation, and Autophagy in RAW 264.7 Murine Macrophage Cell Line Challenged with Si/SiO2 Quantum Dots


Abstract
:1. Introduction
2. Materials and Methods
2.1. Quantum Dots
2.2. Cell Culture
2.3. Cell Viability and Proliferation Assessment
2.4. Cell Lysate Preparation
2.5. Total Protein Quantification
2.6. Evaluation of Lactate Dehydrogenase Activity in the Culture Medium
2.7. Intracellular Superoxide Anion Assessment
2.8. Malondialdehyde Assay
2.9. Enzyme Activity Assessment
2.10. Western Blotting
2.11. Inflammatory Biomarkers Assessment
2.12. Autophagosome Labeling
2.13. Statistical Analysis
3. Results and Discussion
3.1. Cytotoxic Assessment of Si/SiO2 Quantum Dots in RAW 264.7 Cells
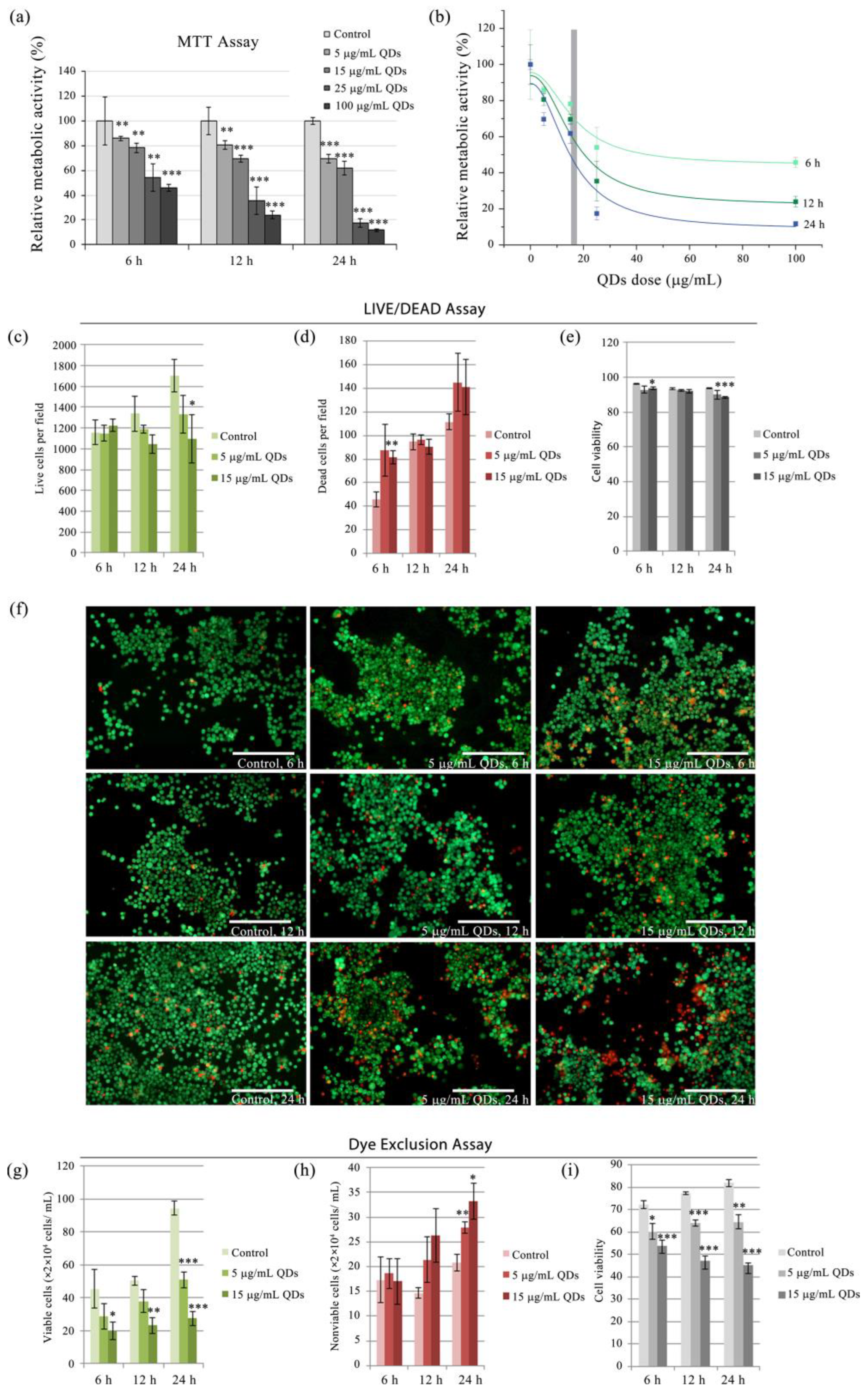
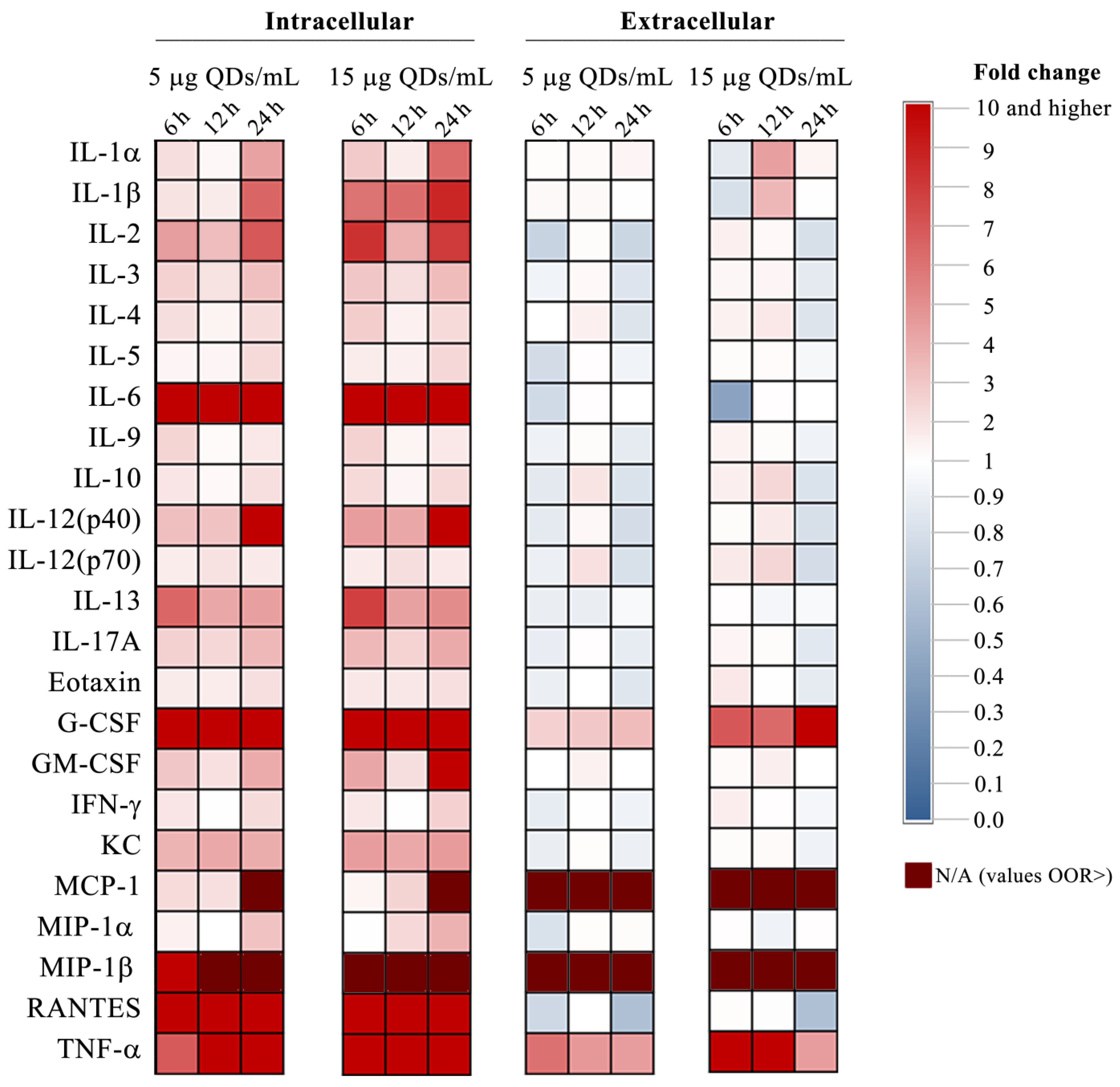
3.2. Impact of Si/SiO2 Quantum Dots on the Inflammatory Response of RAW 264.7 Cells
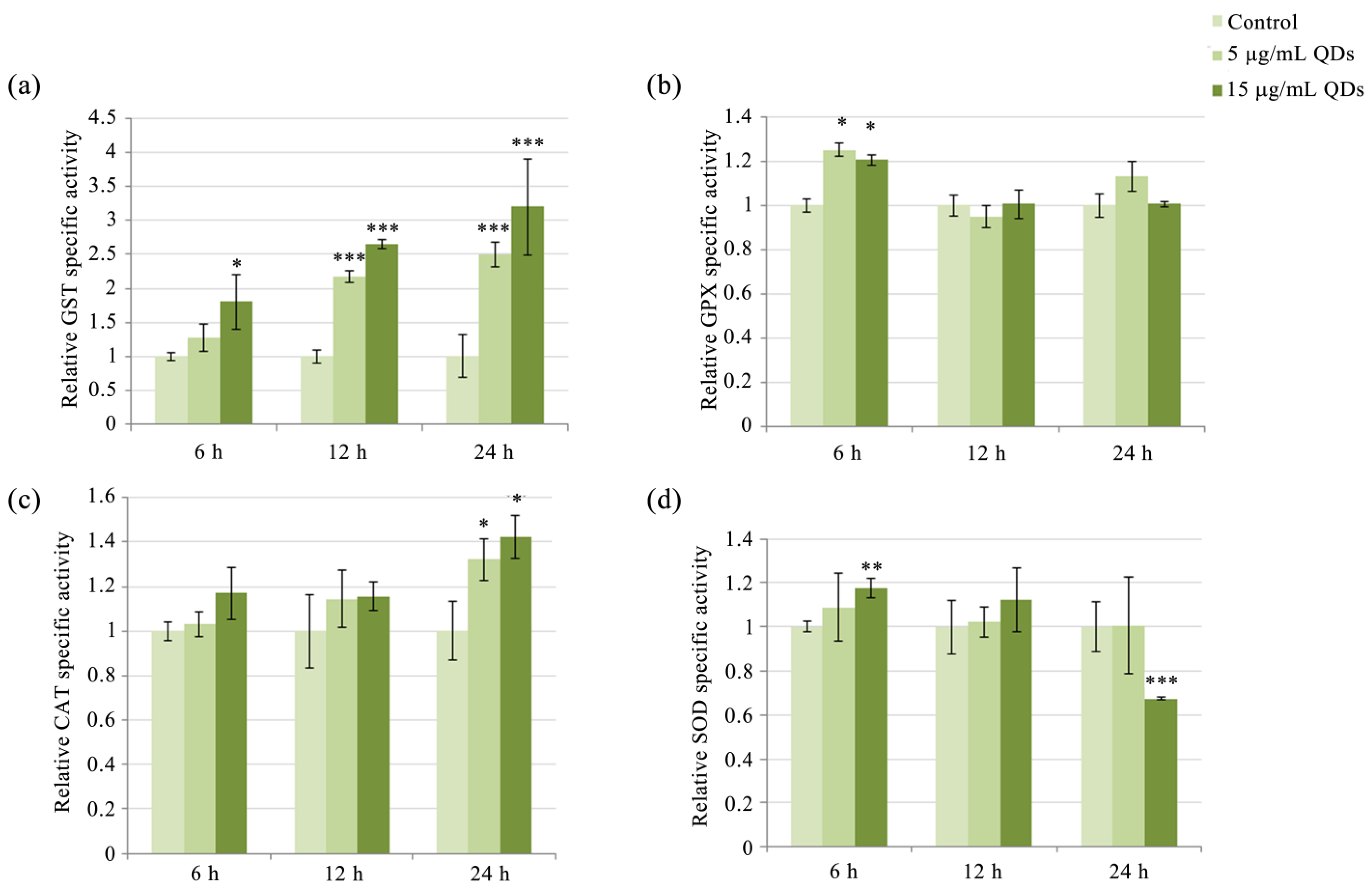
3.3. Modulation of Antioxidative Enzyme Activity in RAW 264.7 Cells Exposed to Si/SiO2 QDs
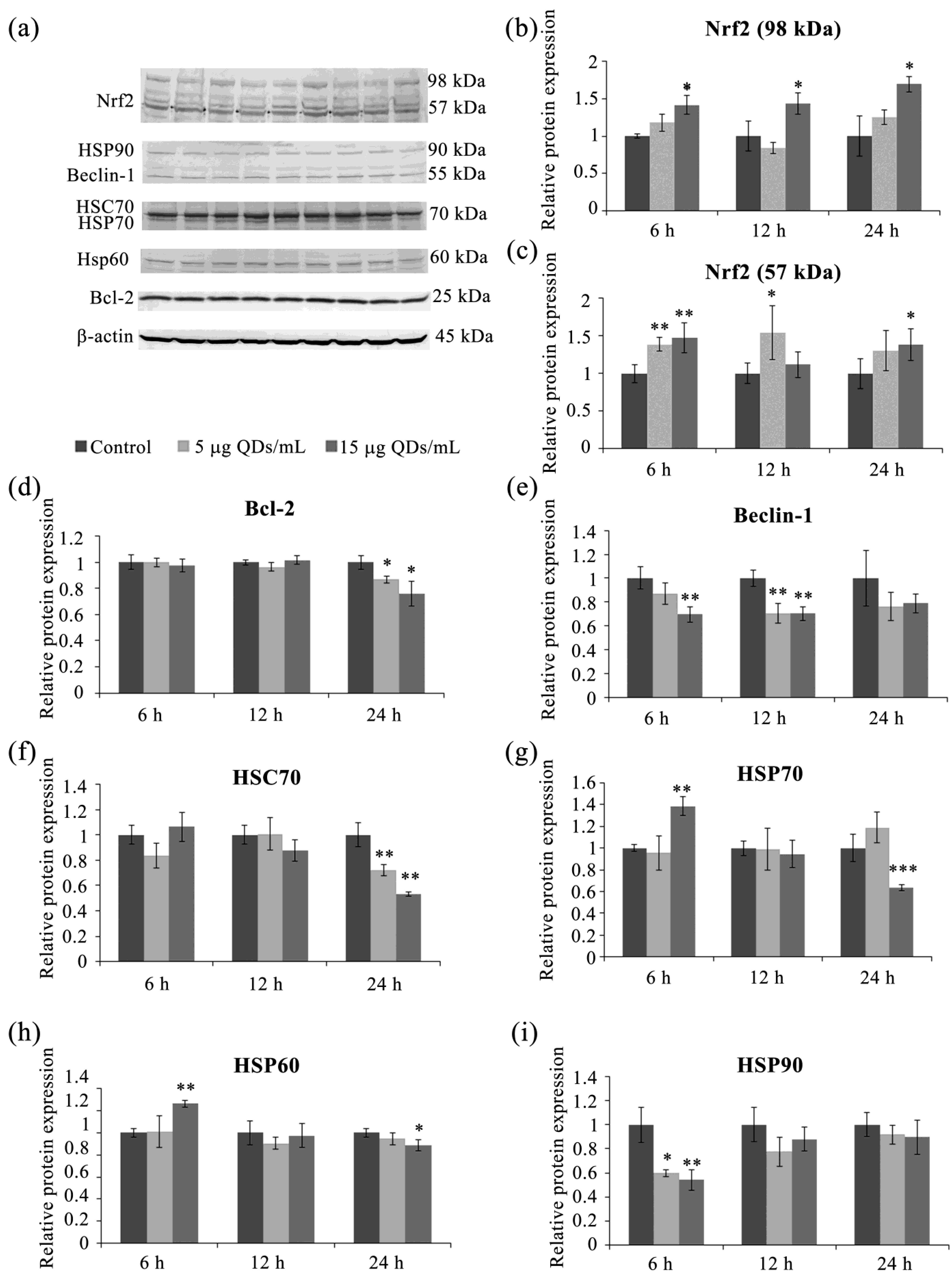
3.4. Key Proteins of the Stress Response Pathways Activated in RAW 264.7 Cells Exposed to Si/SiO2 QDs
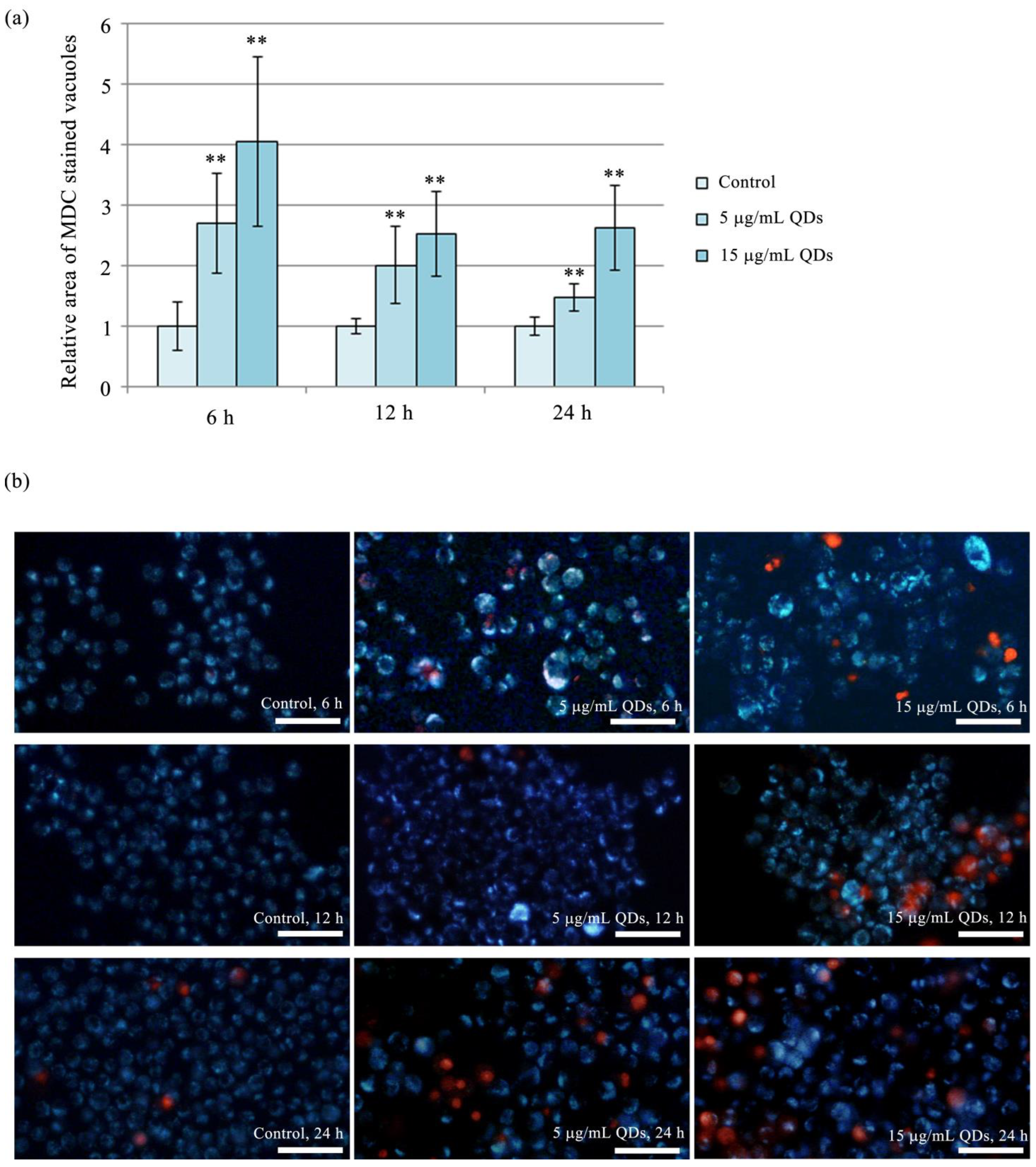
3.5. Autophagy in RAW 264.7 Cells Exposed to Si/SiO2 QDs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bon, P.; Cognet, L. On Some Current Challenges in High-Resolution Optical Bioimaging. ACS Photonics 2022, 9, 2538–2546. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Feng, J.; Zhang, X.; Sun, J.; Fan, C.; Ge, Z. Responsive optical probes for deep-tissue imaging: Photoacoustics and second near-infrared fluorescence. Adv. Drug Deliv. Rev. 2021, 173, 141–163. [Google Scholar] [PubMed]
- Yoon, S.; Cheon, S.Y.; Park, S.; Lee, D.; Lee, Y.; Han, S.; Kim, M.; Koo, H. Recent advances in optical imaging through deep tissue: Imaging probes and techniques. Biomater. Res. 2022, 26, 57. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Peng, F.; Zhong, Y.; Su, Y.; He, Y. Fluorescent quantum dots: Synthesis, biomedical optical imaging, and biosafety assessment. Colloids Surf. B Biointerfaces 2014, 124, 132–139. [Google Scholar]
- Smith, B.R.; Gambhir, S.S. Nanomaterials for In Vivo Imaging. Chem. Rev. 2017, 117, 901–986. [Google Scholar]
- Delille, F.; Pu, Y.; Lequeux, N.; Pons, T. Designing the Surface Chemistry of Inorganic Nanocrystals for Cancer Imaging and Therapy. Cancers 2022, 14, 2456. [Google Scholar] [CrossRef]
- Liu, L.; Miao, Q.; Liang, G. Quantum Dots as Multifunctional Materials for Tumor Imaging and Therapy. Materials 2013, 6, 483–499. [Google Scholar] [CrossRef] [Green Version]
- Dirheimer, L.; Pons, T.; Marchal, F.; Bezdetnaya, L. Quantum Dots Mediated Imaging and Phototherapy in Cancer Spheroid Models: State of the Art and Perspectives. Pharmaceutics 2022, 14, 2136. [Google Scholar] [CrossRef]
- Devi, S.; Kumar, M.; Tiwari, A.; Tiwari, V.; Kaushik, D.; Verma, R.; Bhatt, S.; Sahoo, B.M.; Bhattacharya, T.; Alshehri, S.; et al. Quantum Dots: An Emerging Approach for Cancer Therapy. Front. Mater. 2022, 8, 798440. [Google Scholar]
- Arami, H.; Patel, C.B.; Madsen, S.J.; Dickinson, P.J.; Davis, R.M.; Zeng, Y.; Sturges, B.K.; Woolard, K.D.; Habte, F.G.; Akin, D.; et al. Nanomedicine for Spontaneous Brain Tumors: A Companion Clinical Trial. ACS Nano 2019, 13, 2858–2869. [Google Scholar] [CrossRef]
- Gupta, R.; Xie, H. Nanoparticles in Daily Life: Applications, Toxicity and Regulations. J. Environ. Pathol. Toxicol. Oncol. 2018, 37, 209–230. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Zhang, Q.; Reipa, V.; Wang, N.S.; Stratmeyer, M.; Hitchins, V.; Goering, P. Comparison of cytotoxic and inflammatory responses of photoluminescent silicon nanoparticles with silicon micron-sized particles in RAW 264.7 macrophages. J. Appl. Toxicol. 2009, 29, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.-C.; Tan, S.-H.; Hsiao, Y.-C.; Mutalik, C.; Chen, H.-M.; Yougbaré, S.; Kuo, T.-R. Unveiling the Antibacterial Mechanism of Gold Nanoclusters via In Situ Transmission Electron Microscopy. ACS Sustain. Chem. Eng. 2022, 10, 464–471. [Google Scholar] [CrossRef]
- Donaldson, K.; Stone, V.; Tran, C.L.; Kreyling, W.; Borm, P.J.A. Nanotoxicology. Occup. Environ. Med. 2004, 61, 727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voicu, S.N.; Balas, M.; Stan, M.S.; Trică, B.; Serban, A.I.; Stanca, L.; Hermenean, A.; Dinischiotu, A. Amorphous Silica Nanoparticles Obtained by Laser Ablation Induce Inflammatory Response in Human Lung Fibroblasts. Materials 2019, 12, 1026. [Google Scholar] [CrossRef] [Green Version]
- Herd, H.L.; Bartlett, K.T.; Gustafson, J.A.; McGill, L.D.; Ghandehari, H. Macrophage silica nanoparticle response is phenotypically dependent. Biomaterials 2015, 53, 574–582. [Google Scholar] [CrossRef] [Green Version]
- Stanca, L.; Sima, C.; Voicu, S.; Serban, A.; Dinischiotu, A. In vitro evaluation of the morphological and biochemical changes induced by Si/SiO2 QDs exposure of HepG2 cells. Rom. Rep. Phys. 2015, 67, 1512–1524. [Google Scholar]
- Stan, M.S.; Memet, I.; Sima, C.; Popescu, T.; Teodorescu, V.S.; Hermenean, A.; Dinischiotu, A. Si/SiO2 quantum dots cause cytotoxicity in lung cells through redox homeostasis imbalance. Chem. Biol. Interact. 2014, 220, 102–115. [Google Scholar] [CrossRef]
- Allison, A.C.; Harington, J.S.; Birbeck, M. An examination of the cytotoxic effects of silica on macrophages. J. Exp. Med. 1966, 124, 141–154. [Google Scholar] [CrossRef]
- Yu, Y.; Li, Y.; Wang, W.; Jin, M.; Du, Z.; Li, Y.; Duan, J.; Yu, Y.; Sun, Z. Acute toxicity of amorphous silica nanoparticles in intravenously exposed ICR mice. PLoS ONE 2013, 8, e61346. [Google Scholar] [CrossRef] [Green Version]
- Berghaus, L.J.; Moore, J.N.; Hurley, D.J.; Vandenplas, M.L.; Fortes, B.P.; Wolfert, M.A.; Boons, G.-J. Innate immune responses of primary murine macrophage-lineage cells and RAW 264.7 cells to ligands of Toll-like receptors 2, 3, and 4. Comp. Immunol. Microbiol. Infect. Dis. 2010, 33, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elisia, I.; Pae, H.B.; Lam, V.; Cederberg, R.; Hofs, E.; Krystal, G. Comparison of RAW 264.7, human whole blood and PBMC assays to screen for immunomodulators. J. Immunol. Methods 2018, 452, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Zavala, J.; Freedman, A.N.; Szilagyi, J.T.; Jaspers, I.; Wambaugh, J.F.; Higuchi, M.; Rager, J.E. New Approach Methods to Evaluate Health Risks of Air Pollutants: Critical Design Considerations for In Vitro Exposure Testing. Int. J. Environ. Res. Public Health 2020, 17, 2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farias, I.V.; Fratoni, E.; Theindl, L.C.; de Campos, A.M.; Dalmarco, E.M.; Reginatto, F.H. In Vitro Free Radical Scavenging Properties and Anti-Inflammatory Activity of Ilex paraguariensis (Maté) and the Ability of Its Major Chemical Markers to Inhibit the Production of Proinflammatory Mediators. Mediat. Inflamm. 2021, 2021, 7688153. [Google Scholar] [CrossRef]
- Takahashi, S.; Ferdousi, F.; Yamamoto, S.; Hirano, A.; Nukaga, S.; Nozaki, H.; Isoda, H. Botryococcus terribilis Ethanol Extract Exerts Anti-inflammatory Effects on Murine RAW264 Cells. Int. J. Mol. Sci. 2023, 24, 6666. [Google Scholar] [CrossRef]
- Jalava, P.I.; Aakko-Saksa, P.; Murtonen, T.; Happo, M.S.; Markkanen, A.; Yli-Pirilä, P.; Hakulinen, P.; Hillamo, R.; Mäki-Paakkanen, J.; Salonen, R.O.; et al. Toxicological properties of emission particles from heavy duty engines powered by conventional and bio-based diesel fuels and compressed natural gas. Part. Fibre Toxicol. 2012, 9, 37. [Google Scholar] [CrossRef]
- Deng, H.; Konopka, C.J.; Prabhu, S.; Sarkar, S.; Medina, N.G.; Fayyaz, M.; Arogundade, O.H.; Vidana Gamage, H.E.; Shahoei, S.H.; Nall, D.; et al. Dextran-Mimetic Quantum Dots for Multimodal Macrophage Imaging In Vivo, Ex Vivo, and In Situ. ACS Nano 2022, 16, 1999–2012. [Google Scholar] [CrossRef]
- Petrache, S.N.; Stanca, L.; Serban, A.I.; Sima, C.; Staicu, A.C.; Munteanu, M.C.; Costache, M.; Burlacu, R.; Zarnescu, O.; Dinischiotu, A. Structural and Oxidative Changes in the Kidney of Crucian Carp Induced by Silicon-Based Quantum Dots. Int. J. Mol. Sci. 2012, 13, 10193–10211. [Google Scholar] [CrossRef] [Green Version]
- Grigoriu, C.; Suwa, K.; Muray, K.; Suematsu, H.; Nicolae, I.; Ciupină, V.; Prodan, G. Study of Si/SiO2 nanoparticles produced by laser ablation. In Nanophotonics; SPIE: Bellingham, WA, USA, 2006; Volume 6195, pp. 312–319. [Google Scholar]
- Grigoriu, C.; Nicolae, I.; Ciupina, V.; Prodan, G.; Suematsu, K. Influence of the experimental paramet silicon nanoparticles produced by laser ablation. J. Optoelectron. Adv. Mater. 2004, 6, 825–830. [Google Scholar]
- Stan, M.S.; Sima, C.; Cinteza, L.O.; Dinischiotu, A. Silicon-based quantum dots induce inflammation in human lung cells and disrupt extracellular matrix homeostasis. FEBS J. 2015, 282, 2914–2929. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- ImageJ. Available online: https://imagej.nih.gov/ij (accessed on 1 June 2023).
- Paoletti, F.; Mocali, A. Determination of superoxide dismutase activity by purely chemical system based on NAD(P)H oxidation. Methods Enzym. 1990, 186, 209–220. [Google Scholar]
- Aebi, H. Catalase in vitro. Methods Enzym. 1984, 105, 121–126. [Google Scholar]
- Beutler, E. Red Cell Metabolism; Churchill Livingstone: Edinburgh, Scotland, 1986. [Google Scholar]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974, 249, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Grisham, M.B.; Johnson, G.G.; Lancaster, J.R., Jr. Quantitation of nitrate and nitrite in extracellular fluids. Methods Enzym. 1996, 268, 237–246. [Google Scholar]
- Brenneis, C.; Maier, T.J.; Schmidt, R.; Hofacker, A.; Zulauf, L.; Jakobsson, P.J.; Scholich, K.; Geisslinger, G. Inhibition of prostaglandin E2 synthesis by SC-560 is independent of cyclooxygenase 1 inhibition. FASEB J. 2006, 20, 1352–1360. [Google Scholar] [CrossRef]
- Park, J.Y.; Pillinger, M.H.; Abramson, S.B. Prostaglandin E2 synthesis and secretion: The role of PGE2 synthases. Clin. Immunol. 2006, 119, 229–240. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Vanlangenakker, N.; Vanden Berghe, T.; Vandenabeele, P. Many stimuli pull the necrotic trigger, an overview. Cell Death Differ. 2012, 19, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Berghe, T.V.; Vanlangenakker, N.; Parthoens, E.; Deckers, W.; Devos, M.; Festjens, N.; Guerin, C.J.; Brunk, U.T.; Declercq, W.; Vandenabeele, P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010, 17, 922–930. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Liu, Y.; Li, Z.; Zhu, P.; Zhao, M. Mitochondria Related Cell Death Modalities and Disease. Front. Cell Dev. Biol. 2022, 10, 832356. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Nitric oxide and cell death. Biochim. Et Biophys. Acta (BBA)—Bioenerg. 1999, 1411, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Joosten, L.A.; Smeets, R.L.; Koenders, M.I.; van den Bersselaar, L.A.; Helsen, M.M.; Oppers-Walgreen, B.; Lubberts, E.; Iwakura, Y.; van de Loo, F.A.; van den Berg, W.B. Interleukin-18 promotes joint inflammation and induces interleukin-1-driven cartilage destruction. Am. J. Pathol. 2004, 165, 959–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [Green Version]
- Napierska, D.; Thomassen, L.C.; Lison, D.; Martens, J.A.; Hoet, P.H. The nanosilica hazard: Another variable entity. Part. Fibre Toxicol. 2010, 7, 39. [Google Scholar] [CrossRef] [Green Version]
- Ghiazza, M.; Polimeni, M.; Fenoglio, I.; Gazzano, E.; Ghigo, D.; Fubini, B. Does vitreous silica contradict the toxicity of the crystalline silica paradigm? Chem. Res. Toxicol. 2010, 23, 620–629. [Google Scholar] [CrossRef]
- Ding, M.; Chen, F.; Shi, X.; Yucesoy, B.; Mossman, B.; Vallyathan, V. Diseases caused by silica: Mechanisms of injury and disease development. Int. Immunopharmacol. 2002, 2, 173–182. [Google Scholar] [CrossRef]
- Yang, Y.; Du, X.; Wang, Q.; Liu, J.; Zhang, E.; Sai, L.; Peng, C.; Lavin, M.F.; Yeo, A.J.; Yang, X.; et al. Mechanism of cell death induced by silica nanoparticles in hepatocyte cells is by apoptosis. Int. J. Mol. Med. 2019, 44, 903–912. [Google Scholar] [CrossRef] [Green Version]
- Corbalan, J.J.; Medina, C.; Jacoby, A.; Malinski, T.; Radomski, M.W. Amorphous silica nanoparticles trigger nitric oxide/peroxynitrite imbalance in human endothelial cells: Inflammatory and cytotoxic effects. Int. J. Nanomed. 2011, 6, 2821–2835. [Google Scholar]
- Tilley, S.L.; Coffman, T.M.; Koller, B.H. Mixed messages: Modulation of inflammation and immune responses by prostaglandins and thromboxanes. J. Clin. Investig. 2001, 108, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Lu, B.; Fu, J.; Zhu, X.; Song, E.; Song, Y. Amorphous silica nanoparticles induce inflammation via activation of NLRP3 inflammasome and HMGB1/TLR4/MYD88/NF-kb signaling pathway in HUVEC cells. J. Hazard. Mater. 2021, 404, 124050. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Pétrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [Green Version]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Munder, M.; Mallo, M.; Eichmann, K.; Modolell, M. Murine Macrophages Secrete Interferon γ upon Combined Stimulation with Interleukin (IL)-12 and IL-18: A Novel Pathway of Autocrine Macrophage Activation. J. Exp. Med. 1998, 187, 2103–2108. [Google Scholar] [CrossRef] [Green Version]
- Orosz, L.; Papanicolaou, E.G.; Seprényi, G.; Megyeri, K. IL-17A and IL-17F induce autophagy in RAW 264.7 macrophages. Biomed. Pharmacother. 2016, 77, 129–134. [Google Scholar] [CrossRef]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef]
- Chung, E.Y.; Kim, S.J.; Ma, X.J. Regulation of cytokine production during phagocytosis of apoptotic cells. Cell Res. 2006, 16, 154–161. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.M.C.; Achuthan, A.A.; Hamilton, J.A. GM-CSF: A Promising Target in Inflammation and Autoimmunity. ImmunoTargets Ther. 2020, 9, 225–240. [Google Scholar] [CrossRef]
- Link, H. Current state and future opportunities in granulocyte colony-stimulating factor (G-CSF). Support. Care Cancer 2022, 30, 7067–7077. [Google Scholar] [CrossRef]
- Morrison, R.J.; Singhal, S.S.; Bidani, A.; Heming, T.A.; Awasthi, S. Glutathione S-transferases of rabbit lung macrophages. Toxicol. Appl. Pharmacol. 1998, 148, 229–236. [Google Scholar] [CrossRef]
- Marnett, L.J. Generation of mutagens during arachidonic acid metabolism. Cancer Metastasis Rev. 1994, 13, 303–308. [Google Scholar] [CrossRef]
- Diczfalusy, U.; Falardeau, P.; Hammarström, S. Conversion of prostaglandin endoperoxides to C17-hydroxy acids catalyzed by human platelet thromboxane synthase. FEBS Lett. 1977, 84, 271–274. [Google Scholar] [CrossRef] [Green Version]
- Plastaras, J.P.; Guengerich, F.P.; Nebert, D.W.; Marnett, L.J. Xenobiotic-metabolizing cytochromes P450 convert prostaglandin endoperoxide to hydroxyheptadecatrienoic acid and the mutagen, malondialdehyde. J. Biol. Chem. 2000, 275, 11784–11790. [Google Scholar] [CrossRef] [Green Version]
- Andrés, C.M.; Pérez de la Lastra, J.M.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. Chemistry of Hydrogen Peroxide Formation and Elimination in Mammalian Cells, and Its Role in Various Pathologies. Stresses 2022, 2, 256–274. [Google Scholar] [CrossRef]
- Bukowska, B.; Michałowicz, J.; Pieniazek, D.; Sicińska, P.; Duda, W. Superoxide Dismutases and Their Inhibitors-the Role in Some Diseases. Curr. Enzym. Inhib. 2006, 2, 379–397. [Google Scholar] [CrossRef]
- Alvarez, B.; Demicheli, V.; Durán, R.; Trujillo, M.; Cerveñansky, C.; Freeman, B.A.; Radi, R. Inactivation of human Cu, Zn superoxide dismutase by peroxynitrite and formation of histidinyl radical. Free. Radic. Biol. Med. 2004, 37, 813–822. [Google Scholar] [CrossRef]
- Triboulet, S.; Aude-Garcia, C.; Carrière, M.; Diemer, H.; Proamer, F.; Habert, A.; Chevallet, M.; Collin-Faure, V.; Strub, J.M.; Hanau, D.; et al. Molecular responses of mouse macrophages to copper and copper oxide nanoparticles inferred from proteomic analyses. Mol. Cell. Proteom. 2013, 12, 3108–3122. [Google Scholar] [CrossRef] [Green Version]
- Gupta, G.; Cappellini, F.; Farcal, L.; Gornati, R.; Bernardini, G.; Fadeel, B. Copper oxide nanoparticles trigger macrophage cell death with misfolding of Cu/Zn superoxide dismutase 1 (SOD1). Part. Fibre Toxicol. 2022, 19, 33. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Bai, Y.; Reece, J.M.; Williams, J.; Liu, D.; Freeman, M.L.; Fahl, W.E.; Shugar, D.; Liu, J.; Qu, W.; et al. Molecular mechanism of human Nrf2 activation and degradation: Role of sequential phosphorylation by protein kinase CK2. Free. Radic. Biol. Med. 2007, 42, 1797–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, Y.; Waguri, S.; Sou, Y.-S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [Green Version]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef]
- Ogier-Denis, E.; Codogno, P. Autophagy: A barrier or an adaptive response to cancer. Biochim. Biophys. Acta 2003, 1603, 113–128. [Google Scholar] [CrossRef]
- Gompel, A.; Somaï, S.; Chaouat, M.; Kazem, A.; Kloosterboer, H.J.; Beusman, I.; Forgez, P.; Mimoun, M.; Rostène, W. Hormonal regulation of apoptosis in breast cells and tissues. Steroids 2000, 65, 593–598. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Boya, P.; González-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Métivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef] [Green Version]
- Snyder, C.M.; Shroff, E.H.; Liu, J.; Chandel, N.S. Nitric Oxide Induces Cell Death by Regulating Anti-Apoptotic BCL-2 Family Members. PLoS ONE 2009, 4, e7059. [Google Scholar] [CrossRef] [Green Version]
- Hanagata, N.; Zhuang, F.; Connolly, S.; Li, J.; Ogawa, N.; Xu, M. Molecular responses of human lung epithelial cells to the toxicity of copper oxide nanoparticles inferred from whole genome expression analysis. ACS Nano 2011, 5, 9326–9338. [Google Scholar] [CrossRef] [Green Version]
- Paulo, C.S.; Lino, M.M.; Matos, A.A.; Ferreira, L.S. Differential internalization of amphotericin B--conjugated nanoparticles in human cells and the expression of heat shock protein 70. Biomaterials 2013, 34, 5281–5293. [Google Scholar] [CrossRef]
- Stan, M.S.; Badea, S.; Hermenean, A.; Herman, H.; Trica, B.; Sbarcea, B.G.; Dinischiotu, A. New Insights into the Cell Death Signaling Pathways Triggered by Long-Term Exposure to Silicon-Based Quantum Dots in Human Lung Fibroblasts. Nanomaterials 2021, 11, 323. [Google Scholar] [CrossRef]
- Quintana, F.J.; Cohen, I.R. The HSP60 immune system network. Trends Immunol. 2011, 32, 89–95. [Google Scholar] [CrossRef]
- Ullah, M.; Liu, D.D.; Rai, S.; Concepcion, W.; Thakor, A.S. HSP70-Mediated NLRP3 Inflammasome Suppression Underlies Reversal of Acute Kidney Injury Following Extracellular Vesicle and Focused Ultrasound Combination Therapy. Int. J. Mol. Sci. 2020, 21, 4085. [Google Scholar] [CrossRef]
- Rérole, A.L.; Jego, G.; Garrido, C. Hsp70: Anti-apoptotic and tumorigenic protein. Methods Mol. Biol. 2011, 787, 205–230. [Google Scholar]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Cui, T.; Wang, X. The Interplay Between Autophagy and Regulated Necrosis. Antioxid. Redox Signal. 2022, 38, 550–580. [Google Scholar] [CrossRef]
- Li, W.; Zhao, L.N.; Wei, T.; Zhao, Y.L.; Chen, C.Y. The inhibition of death receptor mediated apoptosis through lysosome stabilization following internalization of carboxyfullerene nanoparticles. Biomaterials 2011, 32, 4030–4041. [Google Scholar] [CrossRef]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef] [Green Version]
- Dice, J.F. Chaperone-mediated autophagy. Autophagy 2007, 3, 295–299. [Google Scholar] [CrossRef] [Green Version]
- Wright, G.S.A. Molecular and pharmacological chaperones for SOD1. Biochem. Soc. Trans. 2020, 48, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Qing, G.; Yan, P.; Xiao, G. Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of IκB kinase (IKK). Cell Res. 2006, 16, 895–901. [Google Scholar] [CrossRef]
- Ambade, A.; Catalano, D.; Lim, A.; Mandrekar, P. Inhibition of heat shock protein (molecular weight 90 kDa) attenuates proinflammatory cytokines and prevents lipopolysaccharide-induced liver injury in mice. Hepatology 2012, 55, 1585–1595. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, A.; Bullock, D.; Lim, A.; Argemi, J.; Orning, P.; Lien, E.; Bataller, R.; Mandrekar, P. Inhibition of HSP90 and Activation of HSF1 Diminish Macrophage NLRP3 Inflammasome Activity in Alcohol-Associated Liver Injury. Alcohol Clin. Exp. Res. 2020, 44, 1300–1311. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Duan, J.; Yu, Y.; Yu, Y.; Li, Y.; Huang, P.; Zhou, X.; Peng, S.; Sun, Z. Silica nanoparticles enhance autophagic activity, disturb endothelial cell homeostasis and impair angiogenesis. Part. Fibre Toxicol. 2014, 11, 50. [Google Scholar] [CrossRef] [Green Version]
- Duan, J.; Yu, Y.; Yu, Y.; Li, Y.; Wang, J.; Geng, W.; Jiang, L.; Li, Q.; Zhou, X.; Sun, Z. Silica nanoparticles induce autophagy and endothelial dysfunction via the PI3K/Akt/mTOR signaling pathway. Int. J. Nanomed. 2014, 9, 5131–5141. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, Y.; Duan, J.; Yang, M.; Yu, Y.; Feng, L.; Yang, X.; Zhou, X.; Zhao, Z.; Sun, Z. Silica nanoparticles induce autophagosome accumulation via activation of the EIF2AK3 and ATF6 UPR pathways in hepatocytes. Autophagy 2018, 14, 1185–1200. [Google Scholar] [CrossRef] [Green Version]
- Petrache Voicu, S.N.; Dinu, D.; Sima, C.; Hermenean, A.; Ardelean, A.; Codrici, E.; Stan, M.S.; Zărnescu, O.; Dinischiotu, A. Silica Nanoparticles Induce Oxidative Stress and Autophagy but Not Apoptosis in the MRC-5 Cell Line. Int. J. Mol. Sci. 2015, 16, 29398–29416. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yu, Y.; Lu, K.; Yang, M.; Li, Y.; Zhou, X.; Sun, Z. Silica nanoparticles induce autophagy dysfunction via lysosomal impairment and inhibition of autophagosome degradation in hepatocytes. Int. J. Nanomed. 2017, 12, 809–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Li, J.; Dong, S.; Cai, X.; Simaiti, A.; Yang, X.; Zhu, X.; Luo, J.; Jiang, L.-H.; Du, B.; et al. Silica nanoparticles induce lung inflammation in mice via ROS/PARP/TRPM2 signaling-mediated lysosome impairment and autophagy dysfunction. Part. Fibre Toxicol. 2020, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Festjens, N.; Vanden Berghe, T.; Vandenabeele, P. Necrosis, a well-orchestrated form of cell demise: Signalling cascades, important mediators and concomitant immune response. Biochim. Biophys. Acta 2006, 1757, 1371–1387. [Google Scholar] [CrossRef] [PubMed]
- Golstein, P.; Kroemer, G. Cell death by necrosis: Towards a molecular definition. Trends Biochem. Sci. 2007, 32, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Jeong, H.; Yu, S.-W. Autophagy as a decisive process for cell death. Exp. Mol. Med. 2020, 52, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E. Cellular targets and mechanisms in the cytotoxic action of non-biodegradable engineered nanoparticles. Curr. Drug Metab. 2013, 14, 976–988. [Google Scholar] [CrossRef]
- Maejima, I.; Takahashi, A.; Omori, H.; Kimura, T.; Takabatake, Y.; Saitoh, T.; Yamamoto, A.; Hamasaki, M.; Noda, T.; Isaka, Y.; et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013, 32, 2336–2347. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.Y.; Lu, J.H. Autophagy and Macrophage Functions: Inflammatory Response and Phagocytosis. Cells 2019, 9, 70. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay Domain | Assay Name | Assay Type |
|---|---|---|
| Cellular proliferation and viability | MTT | Colorimetric |
| LIVE/DEAD | Fluorescence microscopy | |
| Dye exclusion | Microscopy | |
| Stress and membrane integrity | Lactate dehydrogenase activity | Colorimetric |
| Anion superoxide | Colorimetric | |
| Malondialdehyde | Fluorimetric | |
| Antioxidative response | Glutathione-S-transferase activity | Kinetic, spectrofotometric |
| Glutatione peroxidase activity | Kinetic, spectrofotometric | |
| Superoxide dismutase activity | Kinetic, spectrofotometric | |
| Catalase activity | Kinetic, spectrofotometric | |
| Inflammatory response/Inflammatory markers | Cytokines | Conjugated magnetic bead-based immunoassay |
| Cyclooxigenase activity | Colorimetric | |
| Prostaglandin E2 | Enzyme-linked immunoassay | |
| Key proteins of the stress response pathways | Nuclear factor erythroid 2-related factor 2 | Western blot |
| Beclin 1 | Western blot | |
| B-cell lymphoma 2 | Western blot | |
| Heat shock proteins 60, 70, and 90 | Western blot | |
| Caspase 1 activity | Colorimetric | |
| Cellular homeostasis | Autophagy | Fluorescence microscopy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanca, L.; Geicu, O.I.; Serban, A.I.; Dinischiotu, A. Interplay of Oxidative Stress, Inflammation, and Autophagy in RAW 264.7 Murine Macrophage Cell Line Challenged with Si/SiO2 Quantum Dots. Materials 2023, 16, 5083. https://doi.org/10.3390/ma16145083
Stanca L, Geicu OI, Serban AI, Dinischiotu A. Interplay of Oxidative Stress, Inflammation, and Autophagy in RAW 264.7 Murine Macrophage Cell Line Challenged with Si/SiO2 Quantum Dots. Materials. 2023; 16(14):5083. https://doi.org/10.3390/ma16145083
Chicago/Turabian StyleStanca, Loredana, Ovidiu Ionut Geicu, Andreea Iren Serban, and Anca Dinischiotu. 2023. "Interplay of Oxidative Stress, Inflammation, and Autophagy in RAW 264.7 Murine Macrophage Cell Line Challenged with Si/SiO2 Quantum Dots" Materials 16, no. 14: 5083. https://doi.org/10.3390/ma16145083