
Lithium Iron Phosphate and Layered Transition Metal Oxide Cathode for Power Batteries: Attenuation Mechanisms and Modification Strategies

 ,
,
Abstract
:1. Introduction
2. Properties of LFP and NCM

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Battery Properties | Lead–Acid Battery [11] | Nickel–Metal Hydride Battery [12] | Lithium-Ion Battery | ||
|---|---|---|---|---|---|
| LFP [13] | NCM [14] | LCO [15] | |||
| Voltage (V) | 2 | 1.2 | 3.3 | 3.6 | 3.7 |
| Specific energy (Wh·kg−1) | 35–45 | 50–80 | 130–140 | 160–220 | 135–150 |
| Li+ diffusion coefficient (cm2·s−1) | / | / | 10−16–10−14 | 10−11–10−10 | 10−12–10−11 |
| cycle life (times) | 300–500 | 500–1000 | 2000–6000 | 1500–2000 | 500–1000 |
3. Lithium Iron Phosphate (LFP) Battery
3.1. Structure and Properties of LFP
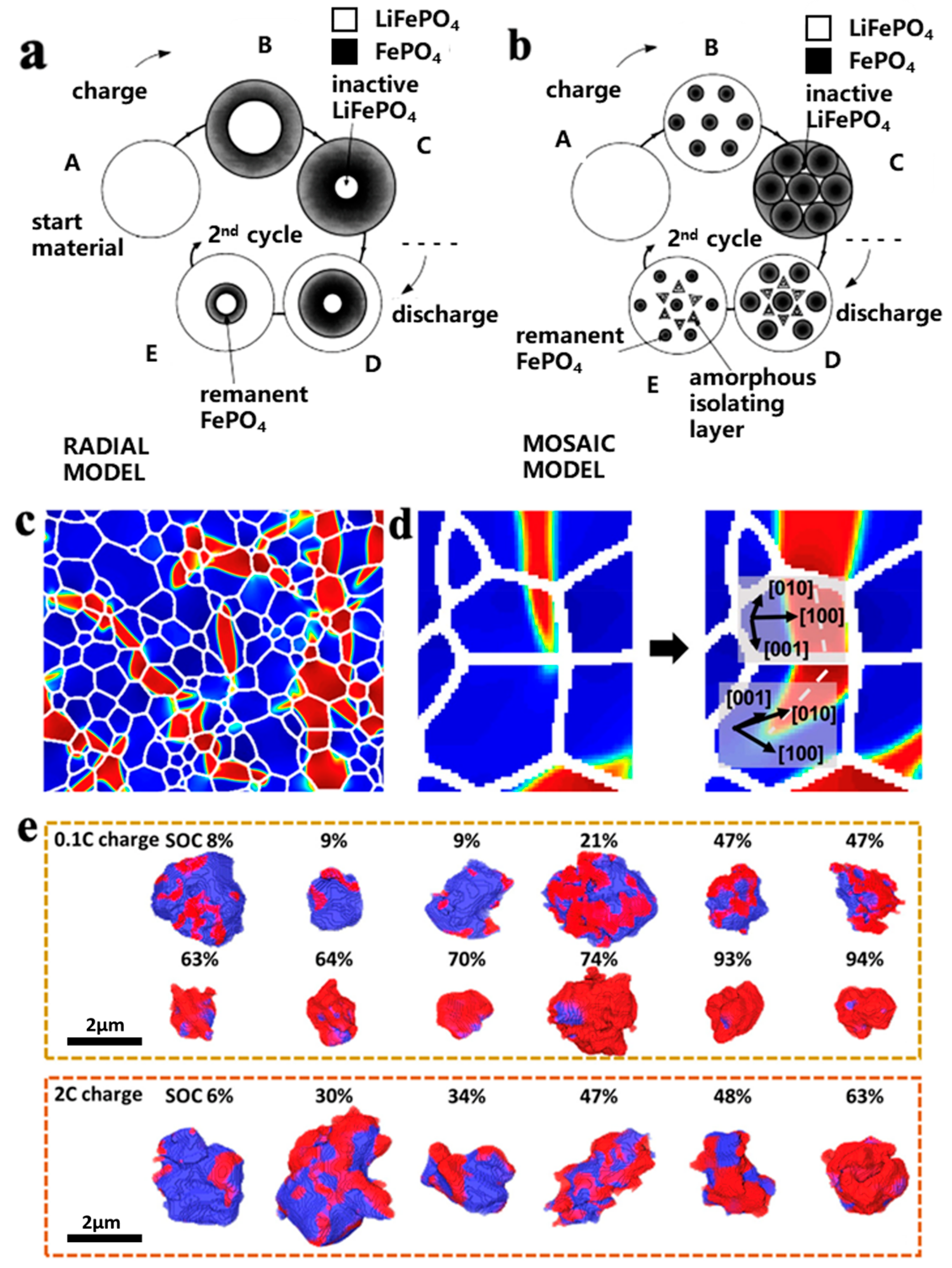
3.2. Life Attenuation Mechanisms of LFP Batteries
3.3. Modification Methods for LFP Batteries
3.3.1. Metal Doping Modification of LFP
3.3.2. Nanosizing and Carbon Coating of LFP Particles
4. NCM Batteries
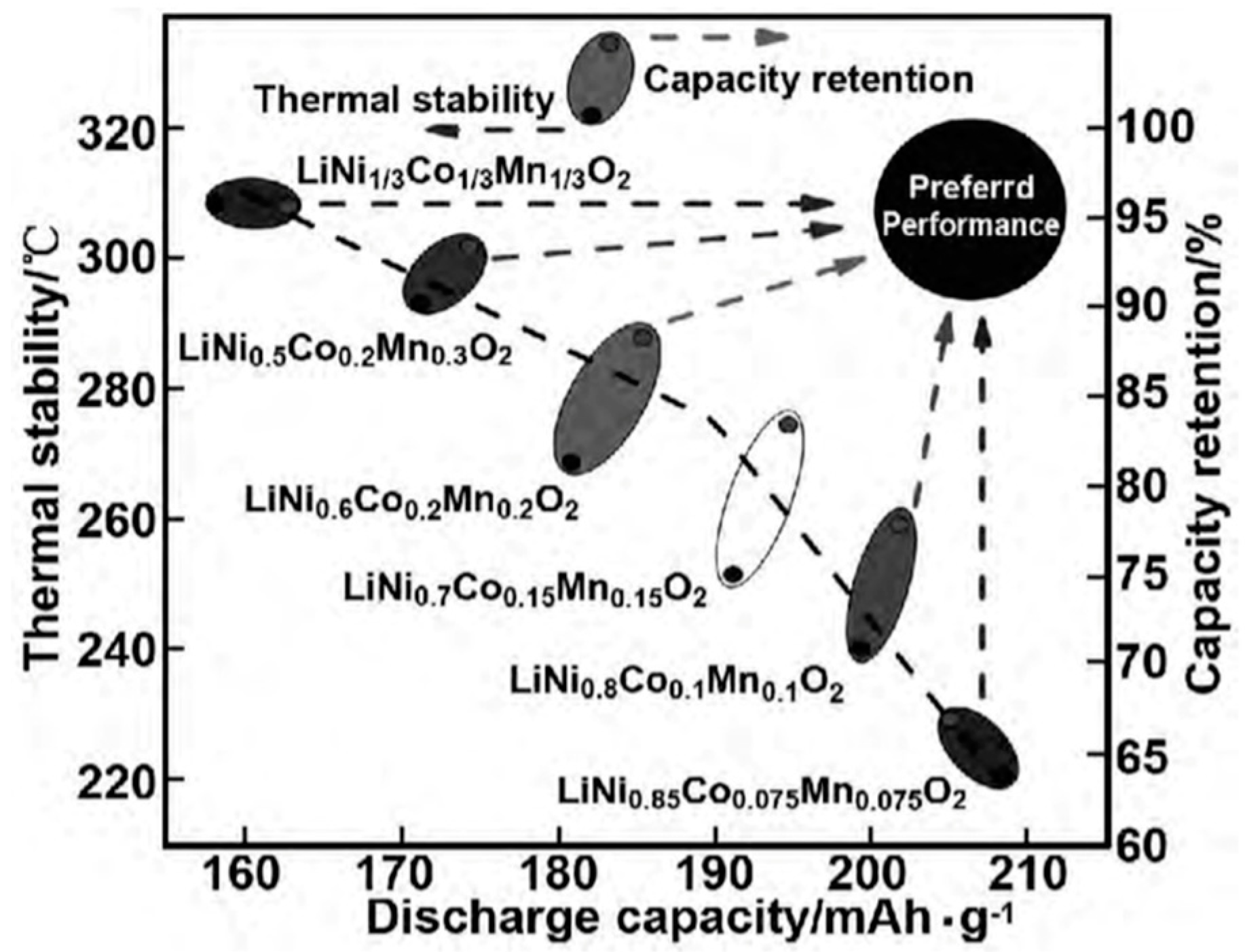
4.1. NCM Structure and Properties
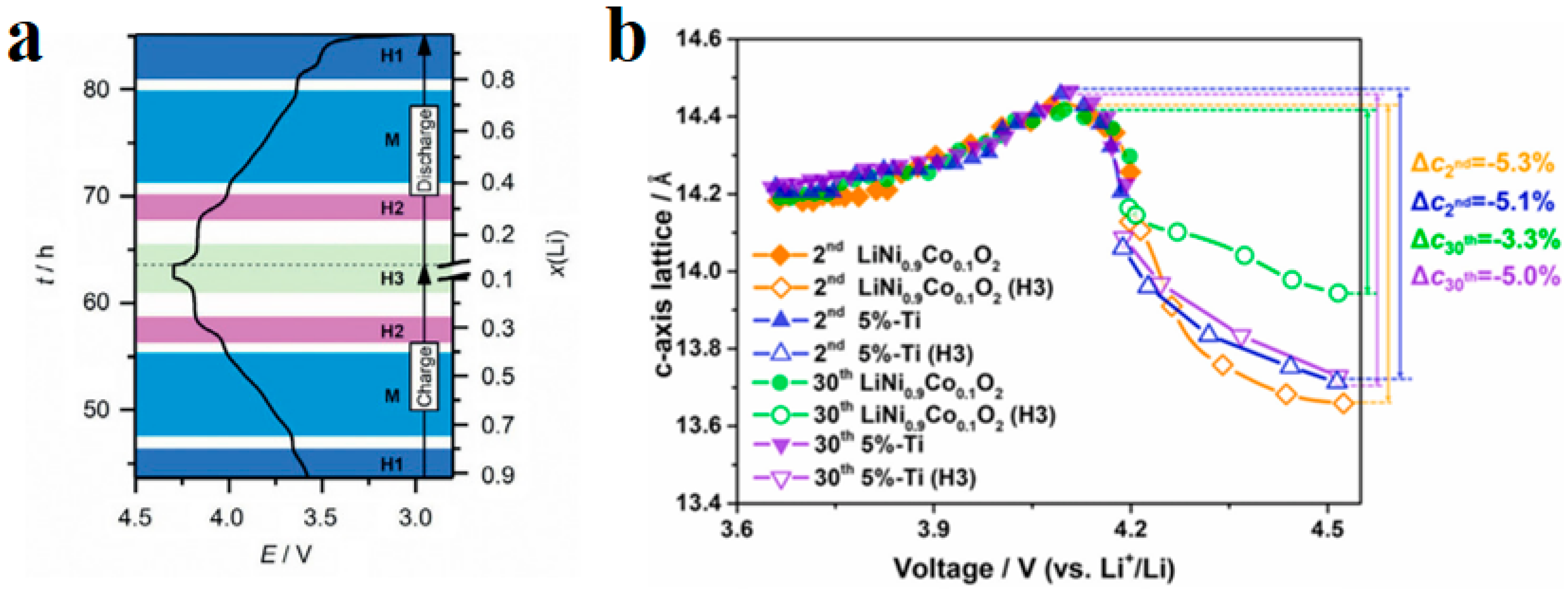
4.2. Life Attenuation Mechanism of NCM Battery
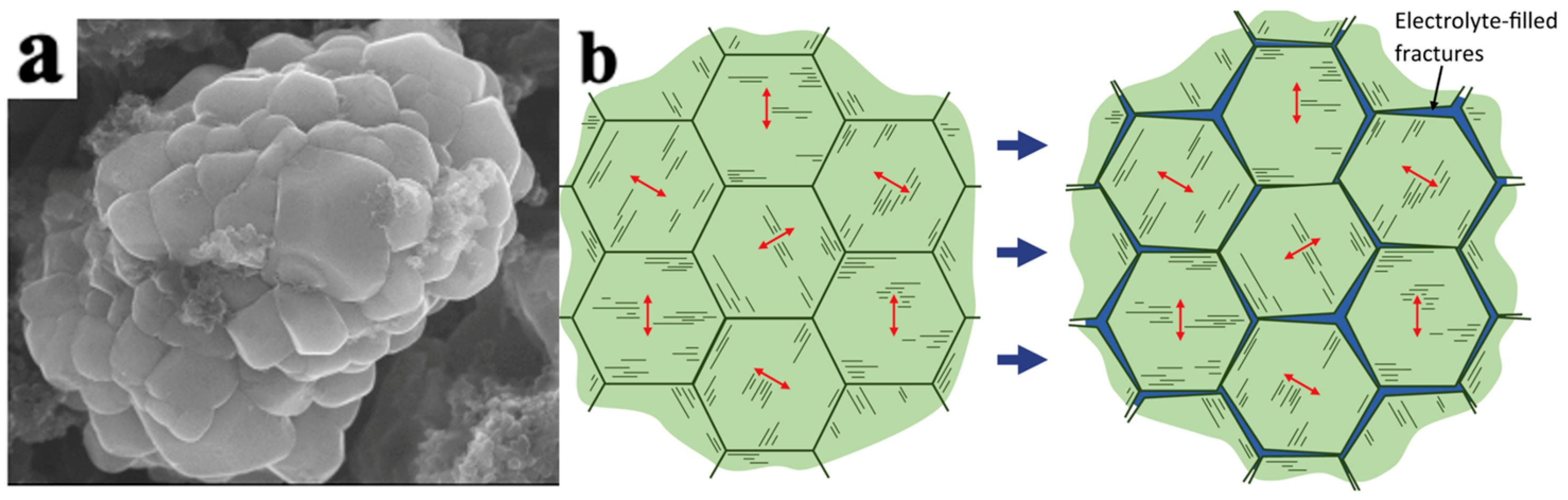
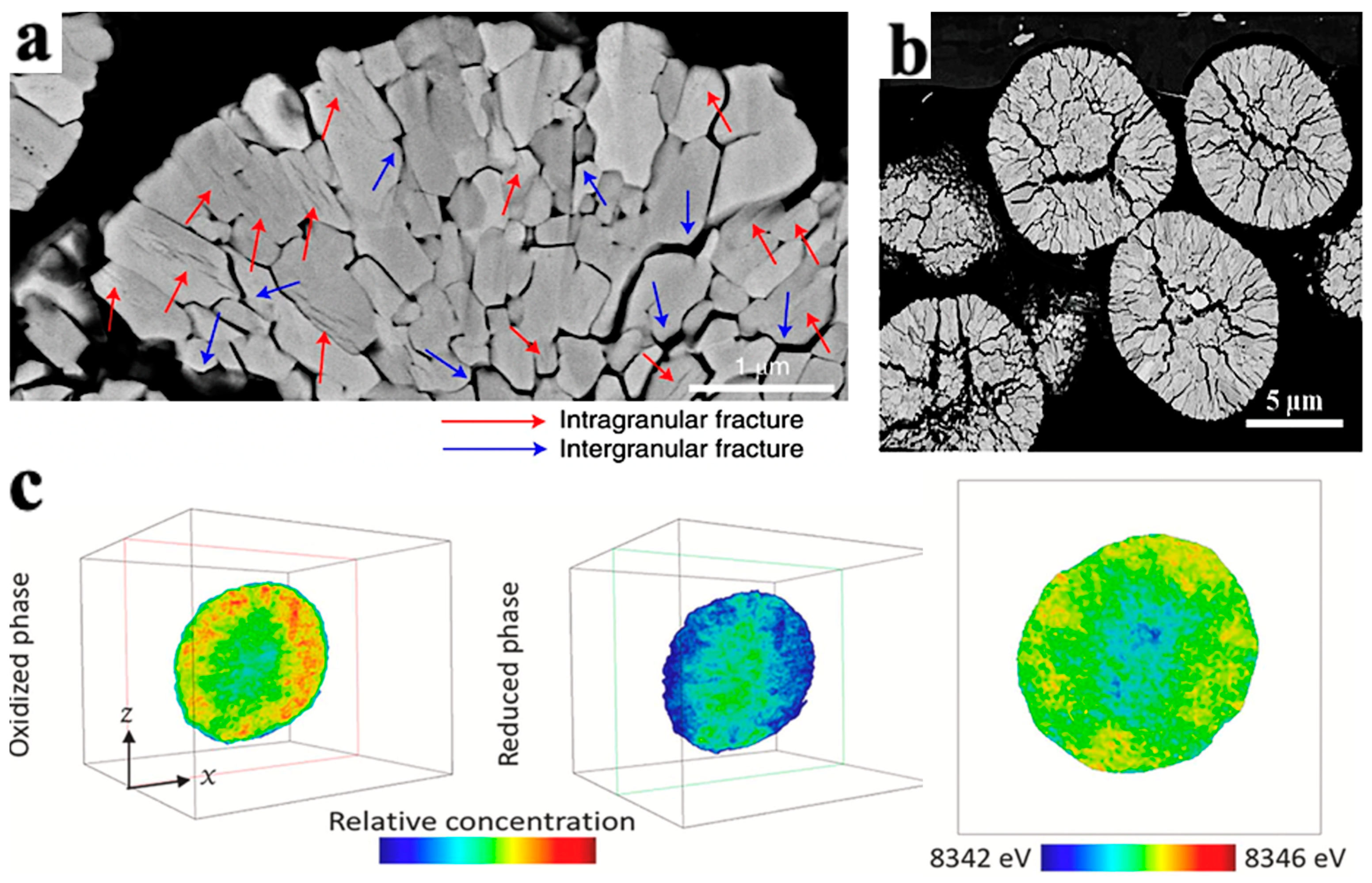
4.2.1. Mechanical Damage of NCM Particles
Intergranular Damage
Intragranular Damage
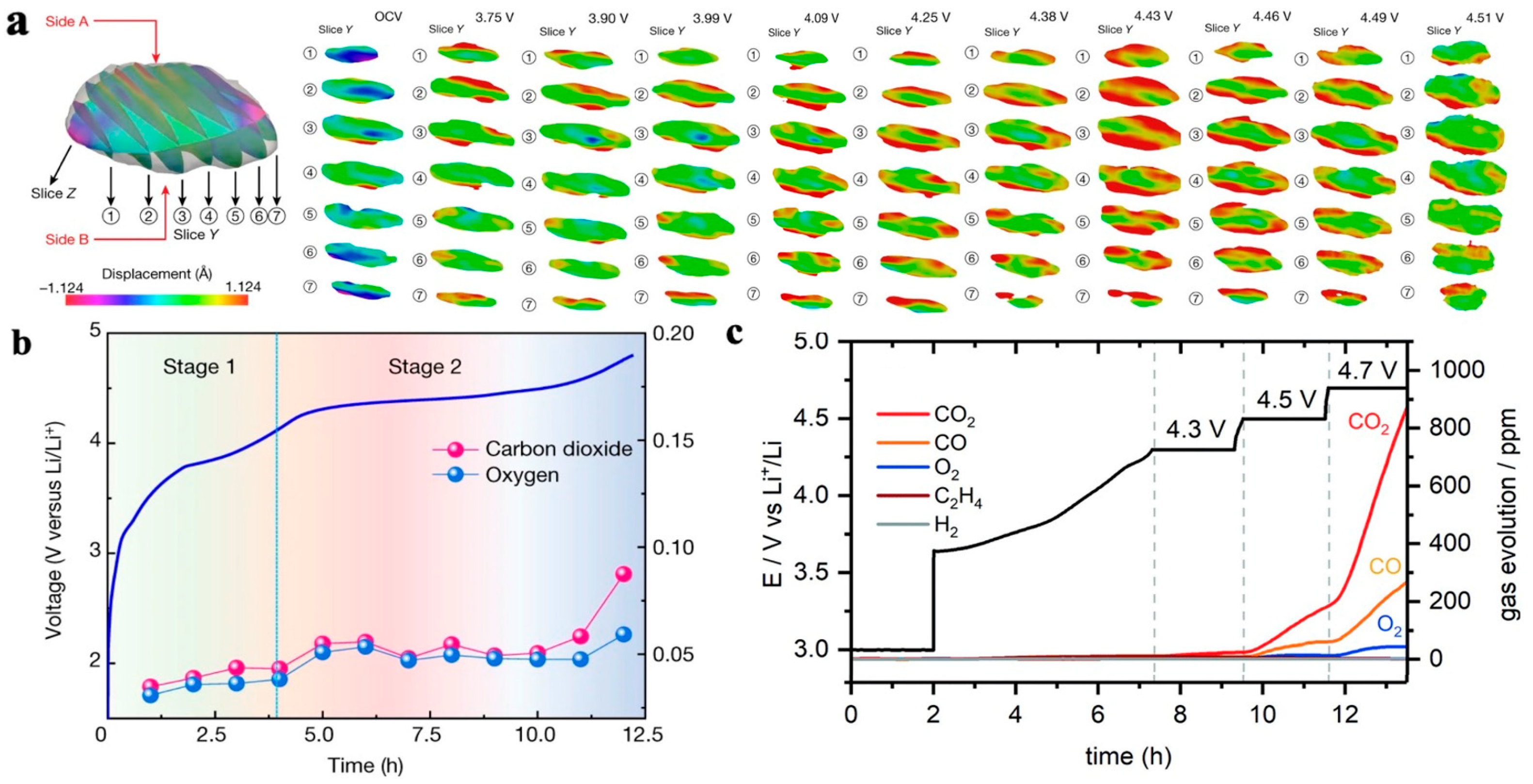
4.2.2. Loss of Lattice Oxygen
4.3. Modification Methods for NCM
4.3.1. Surface Coating Modification
| Morphology | Technique | Cathode Materials | Coating | Capacity (mAh·g−1)/Retention Rate (%) (Rate C/Cycle Times) | Ref. | |
|---|---|---|---|---|---|---|
| Before Coating | After Coating | |||||
Homogeneous coating | Gas phase chemical coating | Li1.2Mn0.54Ni0.13Co0.13O2 | Al2O3 | 251–232/92.43% 0.05 C/30 | 271–257/94.83% 0.05 C/30 | [48] |
| NCM523 | ZrO2 | 216–84/40% 0.03 C/100 | 228–182/83.25% 0.03 C/100 | [49] | ||
| Li1.17Mn0.48Ni0.23Co0.12O2 | MgO | 240–234/97.9% 0.1 C/10 | 260–258/99.55% 0.1 C/10 | [50] | ||
Island coarse coating | Dry coating | LiNi0.815Co0.15Al0.035O2 | Li3PO4 | 195–139/70.55% 1 C/100 | 192–171/89.06% 1 C/100 | [51] |
| NCM525 | Li2MoO4 | 186–97 48% 0.2 C/50 | 178–138 78% 0.2 C/50 | [52] | ||
| Wet coating | Li[Li0.05Ni0.4Co0.15Mn0.4]O2 | Al2O3 | 155–133/86% 1 C/50 | 157–150.7/96% 1 C/50 | [44] | |
| NMC532 | Li3PO4 | 135–44/32.59% 1 C/100 | 214–189/88.32% 1 C/100 | [53] | ||
4.3.2. Elemental Doping Modification of NCM
5. Safety of Power Batteries
6. Conclusions and Outlooks
- The olivine crystal structure of LFP resulted in its low conductivity and ion diffusion rate, leading to the partial deactivation of the cathode particles, a loss of active lithium, and a lower rate performance, limiting the charge and discharge rate in the battery.
- The LFP lithium removal exhibited significant heterogeneity. The FP phase is distributed in a fine filament shape and accompanied by regional condensation, leading to the polarization of the LFP/FP phases in the cathode particles. The uncoordinated polarization behavior between the two phases induced internal stress within the particles, leading to cracks and structural damage in the particles.
- The size of the first-order particles in the NCM materials affected the generation of cracks during their cycling process. When the first-order particle size was smaller than the crack initiation critical size, internal cracks in the first-order particles were hard to initiate and propagate.
- The H2-H3 phase transition can induce cracks in the secondary particles during the cycling of the secondary particles in the NCM materials. The electrolyte may enter the interior of the particles through microcracks and form a passivation film on the surface of the cracks, increasing the volume of the particles and causing breakage of the particles.
- The phase transition of the NCM materials induced lattice oxygen release and structural degradation. In addition, various gases, such as CO2, CO, O2, H2, and C2H4, can be generated, causing safety issues and structural damage.
- The nanosizing and coating of cathode materials need to be applied simultaneously to improve the conductivity and ion diffusion rate and reduce side reactions at the electrode electrolyte interface. For LFP, its interfacial conductivity can be improved through the use of coatings, such as carbon coatings, which show a good coating effect and economic benefits. However, for NCM materials, the coating material needs to serve as a support and physical barrier, requiring careful control of the type and morphology of the coating material. At present, it is still difficult to achieve a thin and uniform coating on the surface of NCM on the basis of low cost, which affects the vibration density and energy density of electrode materials. Further development of coating processes is needed for LFP and NCM to reduce coating costs and increase energy density.
- The main purpose of LFP doping is to improve the conductivity and ion diffusion rate of the material, while the main purpose of NCM material doping is to suppress phase transition. Current research is mostly focused on single-atom doping. Due to the limited performance improvement of single-atom doping, further research is needed on multiple-atom co-doping, elucidating the doping ratios and synergistic effects of multiple doped atoms, and seeking low-cost doping processes.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lyu, Y.; Wu, X.; Wang, K.; Feng, Z.; Cheng, T.; Liu, Y.; Wang, M.; Chen, R.; Xu, L.; Zhou, J.; et al. An overview on the advances of LiCoO2 cathodes for lithium-ion batteries. Adv. Energy Mater. 2021, 11, 2000982. [Google Scholar] [CrossRef]
- Yu, L.; Liu, T.; Amine, R.; Wen, J.; Lu, J.; Amine, K. High nickel and no cobalt-the pursuit of next-generation layered oxide cathodes. Acs Appl. Mater. Inter. 2022, 14, 23056–23065. [Google Scholar] [CrossRef] [PubMed]
- Opitz, A.; Badami, P.; Shen, L.; Vignarooban, K.; Kannan, A.M. Can Li-ion batteries be the panacea for automotive applications? Renew. Sust. Energ. Rev. 2017, 68, 685–692. [Google Scholar] [CrossRef]
- Islam, M.S.; Fisher, C.A.J. Lithium and sodium battery cathode materials: Computational insights into voltage, diffusion and nanostructural properties. Chem. Soc. Rev. 2014, 43, 185–204. [Google Scholar] [CrossRef]
- De Biasi, L.; Schwarz, B.; Brezesinski, T.; Hartmann, P.; Janek, J.; Ehrenberg, H. Chemical, structural, and electronic aspects of formation and degradation behavior on different length scales of ni-rich NCM and Li-rich HE-NCM cathode materials in Li-Ion Batteries. Adv. Mater. 2019, 31, 1900985. [Google Scholar] [CrossRef]
- Ni, J.F.; Su, G.Y.; Zhou, H.H.; Chen, J.T. Study of LiMPO4 as cathode material for lithium ion batteries. Prog. Chem. 2004, 16, 554–560. [Google Scholar]
- Wang, R.; Cui, W.; Chu, F.; Wu, F. Lithium metal anodes: Present and future. J. Energy Chem. 2020, 48, 145–159. [Google Scholar] [CrossRef]
- Yang, C.; Fu, K.; Zhang, Y.; Hitz, E.; Hu, L. Protected lithium-metal anodes in batteries: From liquid to solid. Adv. Mater. 2017, 29, 1701169. [Google Scholar] [CrossRef]
- Zhou, Z.; Jia, X.; Lei, Z. Overview of lithium-ion battery life research. Electromech. Technol. 2019, 7, 117–120. [Google Scholar]
- Marques, P.; Garcia, R.; Kulay, L.; Freire, F. Comparative life cycle assessment of lithium-ion batteries for electric vehicles addressing capacity fade. J. Clean. Prod. 2019, 229, 787–794. [Google Scholar] [CrossRef]
- Vangapally, N.; Rao Penki, T.; Elias, Y.; Muduli, S.; Maddukuri, S.; Luski, S.; Aurbach, D.; Kumar Martha, S. Lead-acid batteries and lead–carbon hybrid systems: A review. J. Power Sources 2023, 579, 233312. [Google Scholar] [CrossRef]
- Cassayre, L.; Gazhov, B.; Zielinski, M.; Biscans, B. Chemical processes for the recovery of valuable metals from spent nickel metal hydride batteries: A review. Renew. Sustain. Energy Rev. 2022, 170, 112983. [Google Scholar] [CrossRef]
- Karuthedath, P.; Pazhaniswamy, S.; Dekanovsky, L.; Balakrishnan, N.; Selvin Paneerselvam, C.; Venkatashamy Reddy, M.; Adams, S.; Sofer, Z. An integrated study on the ionic migration across the nano lithium lanthanum titanate (LLTO) and lithium iron phosphate-carbon (LFP-C) interface in all-solid-state Li-ion batteries. J. Power Sources 2023, 565, 232907. [Google Scholar] [CrossRef]
- Moustafa, M.G.; Sanad Moustafa, M.S. Green fabrication of ZnAl2O4-coated LiFePO4 nanoparticles for enhanced electrochemical performance in Li-ion batteries. J. Alloys Compd. 2022, 903, 163910. [Google Scholar] [CrossRef]
- Sita, L.E.; Pires, D.S.; Faria, L.; Scarminio, J. A simple process to resynthesize the LiCoO2 and LiNi1/3Co1/3Mn1/3O2 compounds from the cathode material extracted from a batch of spent LCO batteries. J. Alloys Compd. 2022, 894, 162350. [Google Scholar] [CrossRef]
- Feng, X.H.; Sun, J.; He, J.; Wei, Y.; Zhou, C.; Sun, R. Research progress of lithium iron phosphate cathode material modification. Energy Storage Sci. Technol. 2022, 11, 467–486. [Google Scholar]
- Sun, T.; Shen, T.; Zheng, Y.; Ren, D.; Zhu, W.; Li, J.; Wang, Y.; Kuang, K.; Rui, X.; Wang, S.; et al. Modeling the inhomogeneous lithium plating in lithium-ion batteries induced by non-uniform temperature distribution. Electrochim. Acta 2022, 425, 140701. [Google Scholar] [CrossRef]
- Yang, L.; Yang, K.; Zheng, J.X.; Xu, K.; Amine, K.; Pan, F. Harnessing the surface structure to enable high-performance cathode materials for lithium-ion batteries. Chem. Soc. Rev. 2020, 49, 4667–4680. [Google Scholar] [CrossRef]
- Padhi, A.K.; Nanjundaswamy, K.S.; Goodenough, J.B. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 1997, 144, 1188. [Google Scholar] [CrossRef]
- Andersson, A.S.; Thomas, J.O. The source of first-cycle capacity loss in LiFePO4. J. Power Sources 2001, 97–98, 498–502. [Google Scholar] [CrossRef]
- Wang, F.; Yang, K.; Ge, M.; Wang, J.; Wang, J.; Xiao, X.; Lee, W.-K.; Li, L.; Tang, M. Reaction heterogeneity in LiFePO4 agglomerates and the role of intercalation-induced stress. ACS Energy Lett. 2022, 7, 1648–1656. [Google Scholar] [CrossRef]
- Guan, Y.; Shen, J.; Wei, X.; Zhu, Q.; Zheng, X.; Zhou, S.; Xu, B. LiFePO4/activated carbon/graphene composite with capacitive-battery characteristics for superior high-rate lithium-ion storage. Electrochim. Acta 2019, 294, 148–155. [Google Scholar] [CrossRef]
- Yan, C.; Wu, K.P.; Jing, P.; Luo, H.; Zhang, Y. Mg-doped porous spherical LiFePO4/C with high tap-density and enhanced electrochemical performance. Mater. Chem. Phys. 2022, 280, 125711. [Google Scholar] [CrossRef]
- Zhang, Q.K.; Xie, X.; Tian, X.J. Research progress of lithium iron phosphate materials for electric vehicles. Battery Ind. 2019, 23, 50–53. [Google Scholar]
- Jiang, H.W.; Liu, Y.W.; Liu, Y.F.; Yue, P.; Zhu, S. Research and application progress of ternary cathode materials for lithium-ion batteries. J. Intraocular Lenses 2018, 47, 2205–2211. [Google Scholar]
- Duan, Y.; Zhang, B.; Zheng, J.; Hu, J.; Wen, J.; Miller, D.J.; Yan, P.; Liu, T.; Guo, H.; Li, W.; et al. Excess Li-ion storage on reconstructed surfaces of nanocrystals to boost battery performance. Nano Lett. 2017, 17, 6018–6026. [Google Scholar] [CrossRef]
- Wu, X.L.; Guo, Y.G.; Su, J.; Xiong, J.W.; Zhang, Y.L.; Wan, L.J. Carbon-nanotube-decorated nano-LiFePO4@C cathode material with superior high-rate and low-temperature performances for lithium-ion batteries. Advanced Energy Materials 2013, 3, 1155–1160. [Google Scholar] [CrossRef]
- Noh, H.-J.; Youn, S.; Yoon, C.S.; Sun, Y.-K. Comparison of the structural and electrochemical properties of layered Li [NixCoyMnz]O2 (x = 1/3, 0.5, 0.6, 0.7, 0.8 and 0.85) cathode material for lithium-ion batteries. J. Power Sources 2013, 233, 121–130. [Google Scholar] [CrossRef]
- Palacín, M.R.; de Guibert, A. Why do batteries fail? Science 2016, 351, 1253292. [Google Scholar] [CrossRef]
- Stallard, J.C.; Wheatcroft, L.; Booth, S.G.; Boston, R.; Corr, S.A.; De Volder, M.F.L.; Inkson, B.J.; Fleck, N.A. Mechanical properties of cathode materials for lithium-ion batteries. Joule 2022, 6, 984–1007. [Google Scholar] [CrossRef]
- Trevisanello, E.; Ruess, R.; Conforto, G.; Richter, F.H.; Janek, J. Polycrystalline and single crystalline NCM cathode materials—Quantifying particle cracking, active surface area, and lithium diffusion. Adv. Energy Mater. 2021, 11, 2003400. [Google Scholar] [CrossRef]
- Brow, R.; Donakowski, A.; Mesnier, A.; Pereira, D.J.; Steirer, K.X.; Santhanagopalan, S.; Manthiram, A. Mechanical pulverization of Co-free nickel-rich cathodes for improved high-voltage cycling of lithium-ion batteries. ACS Appl. Energy Mater. 2022, 5, 6996. [Google Scholar] [CrossRef]
- Xu, C.; Reeves, P.J.; Jacquet, Q.; Grey, C.P. Phase behavior during electrochemical cycling of Ni-rich cathode materials for Li-ion batteries. Adv. Energy Mater. 2021, 11, 2003404. [Google Scholar] [CrossRef]
- Wu, F.; Liu, N.; Chen, L.; Su, Y.; Tan, G.; Bao, L.; Tan, J.; Zhang, Q.; Lu, Y.; Wang, J.; et al. Improving the reversibility of the H2-H3 phase transitions for layered Ni-rich oxide cathode towards retarded structural transition and enhanced cycle stability. Nano Energy 2019, 59, 50–57. [Google Scholar] [CrossRef]
- Kim, U.H.; Park, G.T.; Conlin, P.; Ashburn, N.; Cho, K.; Yu, Y.-S.; Shapiro, D.A.; Maglia, F.; Kim, S.-J.; Lamp, P.; et al. Cation ordered Ni-rich layered cathode for ultra-long battery life. Energy Environ. Sci. 2021, 14, 1573–1583. [Google Scholar] [CrossRef]
- Yin, S.; Deng, W.T.; Chen, J.; Gao, X.; Zou, G.; Hou, H.; Ji, X. Fundamental and solutions of microcrack in Ni-rich layered oxide cathode materials of lithium-ion batteries. Nano Energy 2021, 83, 105854. [Google Scholar] [CrossRef]
- Park, G.T.; Namkoong, B.; Kim, S.B.; Liu, J.; Yoon, C.S.; Sun, Y.-K. Introducing high-valence elements into cobalt-free layered cathodes for practical lithium-ion batteries. Nat. Energy 2022, 53, 1254. [Google Scholar] [CrossRef]
- Namkoong, B.; Park, N.Y.; Park, G.T.; Shin, J.-Y.; Beierling, T.; Yoon, C.S.; Sun, Y.-K. High-energy Ni-rich cathode materials for long-range and long-life electric vehicles. Adv. Energy Mater. 2022, 12, 2200615. [Google Scholar] [CrossRef]
- Bi, Y.; Tao, J.H.; Wu, Y.Q.; Li, L.; Xu, Y.; Hu, E.; Wu, B.; Hu, J.; Wang, C.; Zhang, J.-G.; et al. Reversible planar gliding and microcracking in a single-crystalline Ni-rich cathode. Science 2020, 370, 1313–1317. [Google Scholar] [CrossRef]
- Liu, T.; Liu, J.J.; Li, L.X.; Yu, L.; Diao, J.; Zhou, T.; Li, S.; Dai, A.; Zhao, W.; Xu, S.; et al. Origin of structural degradation in Li-rich layered oxide cathode. Nature 2022, 606, 305–312. [Google Scholar] [CrossRef]
- Rinkel, B.L.D.; Vivek, J.P.; Garcia-Araez, N.; Grey, C.P. Two electrolyte decomposition pathways at nickel-rich cathode surfaces in lithium-ion batteries. Energy Environ. Sci. 2022, 15, 3416–3438. [Google Scholar] [CrossRef] [PubMed]
- Rinkel, B.L.D.; Vivek, J.P.; Garcia-Araez, N.; Grey, C.P. Two electrolyte decomposition pathways at NMC electrodes in lithium-ion batteries. In ECS Meeting Abstracts; The Electrochemical Society, Inc.: Pennington, NJ, USA, 2022; Volume 241, p. 348. [Google Scholar]
- Akhilash, M.; Salini, P.S.; John, B.; Sujatha, S.; Mercy, T.D. Surface modification on nickel rich cathode materials for lithium-ion cells: A mini review. Chem. Rec. 2023, e202300132. [Google Scholar] [CrossRef] [PubMed]
- Ahaliabadeh, Z.; Kong, X.Z.; Fedorovskaya, E.; Kallio, T. Extensive comparison of doping and coating strategies for Ni-rich positive electrode materials. J. Power Sources 2022, 540, 231633. [Google Scholar] [CrossRef]
- Sanad Moustafa, M.S.; Meselhy Neama, K.; El-Boraey Hanaa, A. Surface protection of NMC811 cathode material via ZnSnO3 perovskite film for enhanced electrochemical performance in rechargeable Li-ion batteries. Colloids Surf. A Physicochem. Eng. Asp. 2023, 672, 131748. [Google Scholar] [CrossRef]
- Sanad Moustafa, M.S.; Meselhy Neama, K.; El-Boraey Hanaa, A.; Toghan, A. Controllable engineering of new ZnAl2O4-decorated LiNi0·8Mn0·1Co0·1O2 cathode materials for high performance lithium-ion batteries. J. Mater. Res. Technol. 2023, 23, 1528–1542. [Google Scholar] [CrossRef]
- Nisar, U.; Muralidharan, N.; Amin, R.; Amin, R.; Belharouak, I. Valuation of surface coatings in high-energy density lithium-ion battery cathode materials. Energy Storage Mater. 2021, 38, 309–328. [Google Scholar] [CrossRef]
- Liu, J.; Manthiram, A. Functional surface modifications of a high capacity layered Li[Li0.2Mn0.54Ni0.13Co0.13]O2 cathode. J. Mater. Chem. 2010, 20, 3961–3967. [Google Scholar] [CrossRef]
- Li, X.; Zhang, K.J.; Mitlin, D.; Yang, Z.Z.; Wang, M.; Tang, Y.; Jiang, F.; Du, Y.; Zheng, J. Fundamental insight into Zr modification of Li- and Mn-rich cathodes: Combined transmission electron microscopy and electrochemical impedance spectroscopy study. Chem. Mater. 2018, 30, 2566–2573. [Google Scholar] [CrossRef]
- Han, E.S.; Li, Y.P.; Zhu, L.Z.; Zhao, Z.L. The effect of MgO coating on Li1. 17Mn0. 48Ni0. 23Co0. 12O2 cathode material for lithium ion batteries. Solid. State Ion. 2014, 255, 113–119. [Google Scholar] [CrossRef]
- Zou, P.; Lin, Z.H.; Fan, M.; Wang, F.; Liu, Y.; Xiong, X.H. Facile and efficient fabrication of Li3PO4-coated Ni-rich cathode for high-performance lithium-ion battery. Appl. Surf. Sci. 2020, 504, 144506. [Google Scholar] [CrossRef]
- Lu, J.; Zhan, C.; Wu, T.P.; Wen, J.G.; Lei, Y.; Jeremy Kropf, A.; Wu, H.M.; Miller Dean, J.; Elam Jeffrey, W.; Sun, Y.K.; et al. Effectively suppressing dissolution of manganese from spinel lithium manganate via a nanoscale surface-doping approach. Nat. Commun. 2014, 5, 5693. [Google Scholar] [CrossRef]
- Sahni, K.; Ashuri, M.; He, Q.R.; Sahore, R.; Bloom Ira, D.; Liu, Y.Z.; Kaduk James, A.; Shaw Leon, L. H3PO4 treatment to enhance the electrochemical properties of Li(Ni1/3Mn1/3Co1/3)O2 and Li(Ni0.5Mn0.3Co0.2)O2 cathodes. Electrochim. Acta 2019, 301, 8–22. [Google Scholar] [CrossRef]
- Dong, S.D.; Zhou, Y.; Hai, C.X. Research progress on modification of Nickel-cobalt-manganese ternary cathode materials for lithium-ion batteries. Polym. Bull. 2018, 8, 112–118. [Google Scholar]
- Wang, J.L.; Nie, Y.; Miao, C.; Tan, Y.; Wen, M.Y.; Xiao, W. Enhanced electrochemical properties of Ni-rich layered cathode materials via Mg2+ and Ti4+ co-doping for lithium-ion batteries. J. Colloid. Interf. Sci. 2021, 601, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Li, P.Y.; Huang, Y.D.; Tang, L.B.; Wei, H.X.; Fu, H.; He, Z.J.; Zheng, J.C. W-Doped LiNi1/3Co1/3Mn1/3O2 with Excellent High-Rate Performance Synthesized via Hydrothermal Lithiation. J. Electrochem. Soc. 2022, 169, 050509. [Google Scholar] [CrossRef]
- Jeevanantham, B.; Shobana, M.K. Enhanced cathode materials for advanced lithium-ion batteries using nickel-rich and lithium/manganese-rich LiNixMnyCozO2. J. Energy Storage 2022, 54, 105353. [Google Scholar]
- Jiang, L.; Wang, J.; Zou, X. Analysis of GB 38031—2020 safety requirements of traction battery used by electric road vehicles. Batt Bimonth. 2020, 50, 276–279. [Google Scholar]
- Bates Alex, M.; Preger, Y.; Torres-Castro, L.; Harrison Katharine, L.; Harris Stephen, J.; Hewson, J. Are solid-state batteries safer than lithium-ion batteries? Joule 2022, 6, 742–755. [Google Scholar] [CrossRef]
- Bak, S.M.; Hu, E.Y.; Zhou, Y.N.; Yu, X.Q.; Senanayake Sanjaya, D.; Cho, S.J.; Kim, K.B.; Chung Kyung, Y.; Yang, X.Q.; Nam, K.W. Structural changes and thermal stability of charged LiNixMnyCozO2 cathode materials studied by combined in situ time-resolved XRD and mass spectroscopy. Acs Appl. Mater. Inter. 2014, 6, 22594–22601. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, D.D.S.; Tenorio, J.A.S.; Botelho, A.B.; Espinosa, D.C.R. Circular recycling strategies for LFP batteries: A review focusing on hydrometallurgy sustainable processing. Metals 2023, 13, 543. [Google Scholar] [CrossRef]
- Liu, K.; Liu, Y.Y.; Lin, D.C.; Pei, A.; Cui, Y. Materials for lithium-ion battery safety. Sci. Adv. 2018, 4, eaas9820. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chen, R.; Gan, L.; Li, H.; Chen, L.Q. Battery safety: From lithium-ion to solid-state batteries. Engineering 2023, 21, 9–14. [Google Scholar] [CrossRef]
- Jung, S.H.; Kim, U.H.; Kim, J.H.; Jun, S.G.; Yoon Chong, S.; Jung Yoon, S.; Sun, Y.K. Ni-rich layered cathode materials with electrochemo-mechanically compliant microstructures for all-solid-state Li batteries. Adv. Energy Mater. 2020, 10, 1903360. [Google Scholar] [CrossRef]
- Deng, S.; Sun, Y.; Li, X.; Ren, Z.H.; Liang, J.W.; Doyle-Davis, K.; Liang, J.N.; Li, W.H.; Banis Mohammad, N.; Sun, Q.; et al. Eliminating the detrimental effects of conductive agents in sulfide-based solid-state batteries. Acs Energy Lett. 2020, 5, 1243–1251. [Google Scholar] [CrossRef]
- Liang, J.W.; Zhu, Y.; Li, X.; Luo, J.; Deng, S.X.; Zhao, Y.; Sun, Y.P.; Wu, D.J.; Hu, Y.F.; Li, W.H.; et al. A gradient oxy-thiophosphate-coated Ni-rich layered oxide cathode for stable all-solid-state Li-ion batteries. Nat. Commun. 2023, 14, 146. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Li, X.J.; Luo, L.J.; He, S.N.; Chen, J.; Liu, Y.F.; Pan, H.G.; Song, Y.; Hu, R.Z. Practical Application of All-solid-state lithium batteries based on high-voltage cathodes: Challenges and progress. Adv. Energy Mater. 2023, 2301230. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, G.; Li, M.; Ye, Z.; Chen, T.; Cao, J.; Yang, H.; Ma, C.; Jia, Z.; Xie, J.; Cui, N.; et al. Lithium Iron Phosphate and Layered Transition Metal Oxide Cathode for Power Batteries: Attenuation Mechanisms and Modification Strategies. Materials 2023, 16, 5769. https://doi.org/10.3390/ma16175769
Zhang G, Li M, Ye Z, Chen T, Cao J, Yang H, Ma C, Jia Z, Xie J, Cui N, et al. Lithium Iron Phosphate and Layered Transition Metal Oxide Cathode for Power Batteries: Attenuation Mechanisms and Modification Strategies. Materials. 2023; 16(17):5769. https://doi.org/10.3390/ma16175769
Chicago/Turabian StyleZhang, Guanhua, Min Li, Zimu Ye, Tieren Chen, Jiawei Cao, Hongbo Yang, Chengbo Ma, Zhenggang Jia, Jiwei Xie, Ning Cui, and et al. 2023. "Lithium Iron Phosphate and Layered Transition Metal Oxide Cathode for Power Batteries: Attenuation Mechanisms and Modification Strategies" Materials 16, no. 17: 5769. https://doi.org/10.3390/ma16175769
APA StyleZhang, G., Li, M., Ye, Z., Chen, T., Cao, J., Yang, H., Ma, C., Jia, Z., Xie, J., Cui, N., & Xiong, Y. (2023). Lithium Iron Phosphate and Layered Transition Metal Oxide Cathode for Power Batteries: Attenuation Mechanisms and Modification Strategies. Materials, 16(17), 5769. https://doi.org/10.3390/ma16175769