
Comparison of TiO2 and ZnO for Heterogeneous Photocatalytic Activation of the Peroxydisulfate Ion in Trimethoprim Degradation
 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Photoreactors and Experimental Parameters
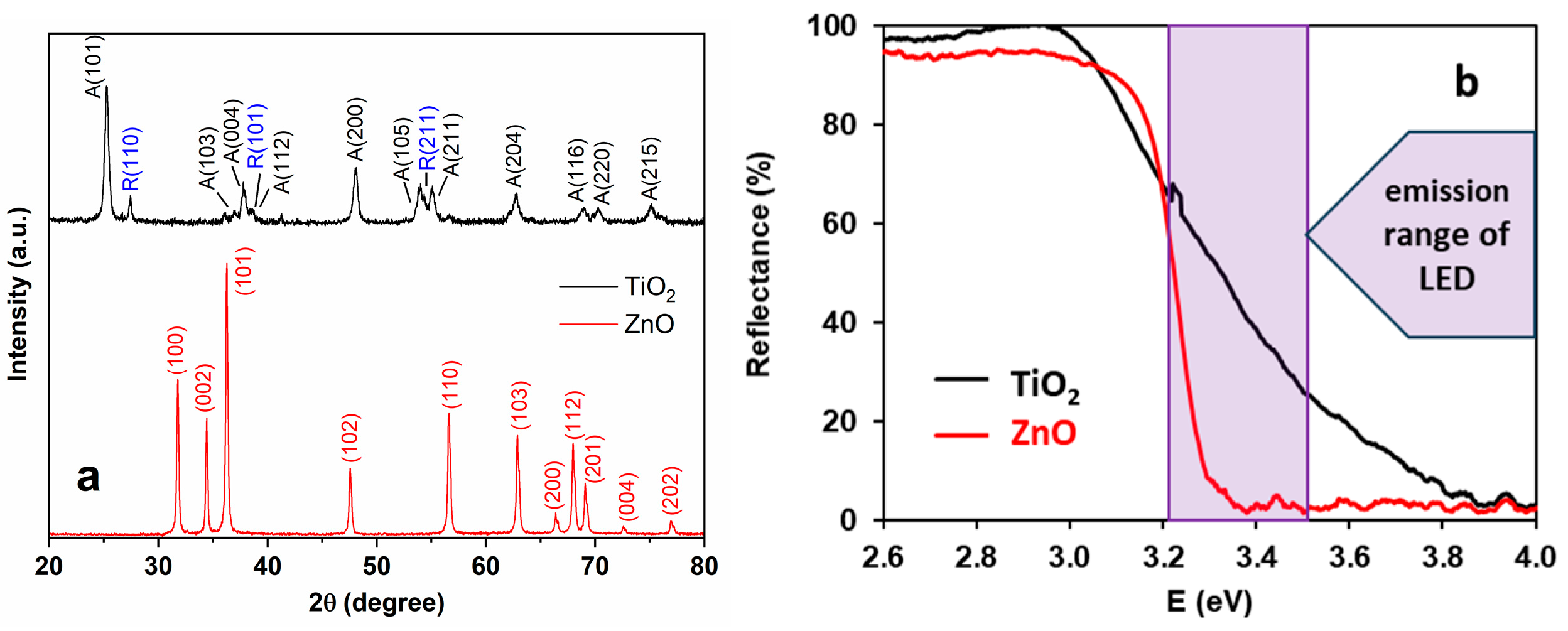
2.3. Characterization of the Light Source
2.4. Characterization of the Photocatalysts
2.5. Analytical Methods
3. Results
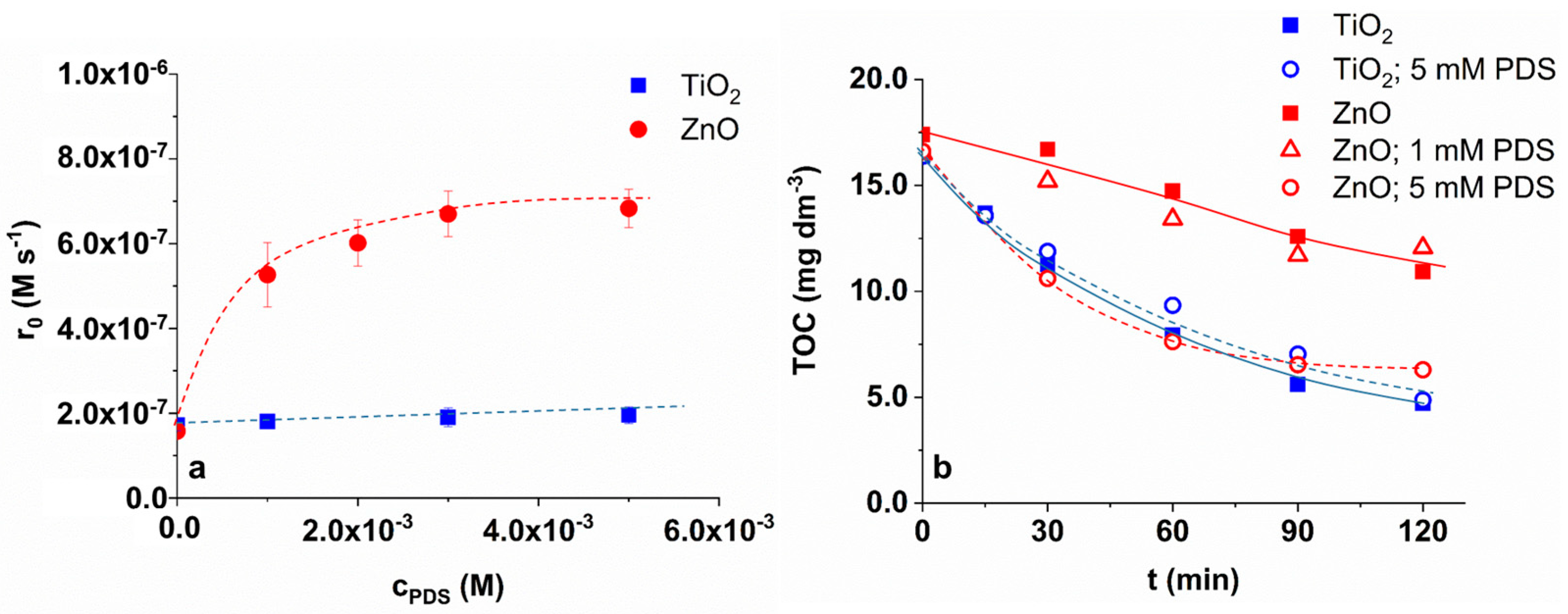
3.1. Effect of PDS and Dissolved O2 on the TRIM Transformation
3.2. Effect of Dissolved O2 on TRIM Transformation
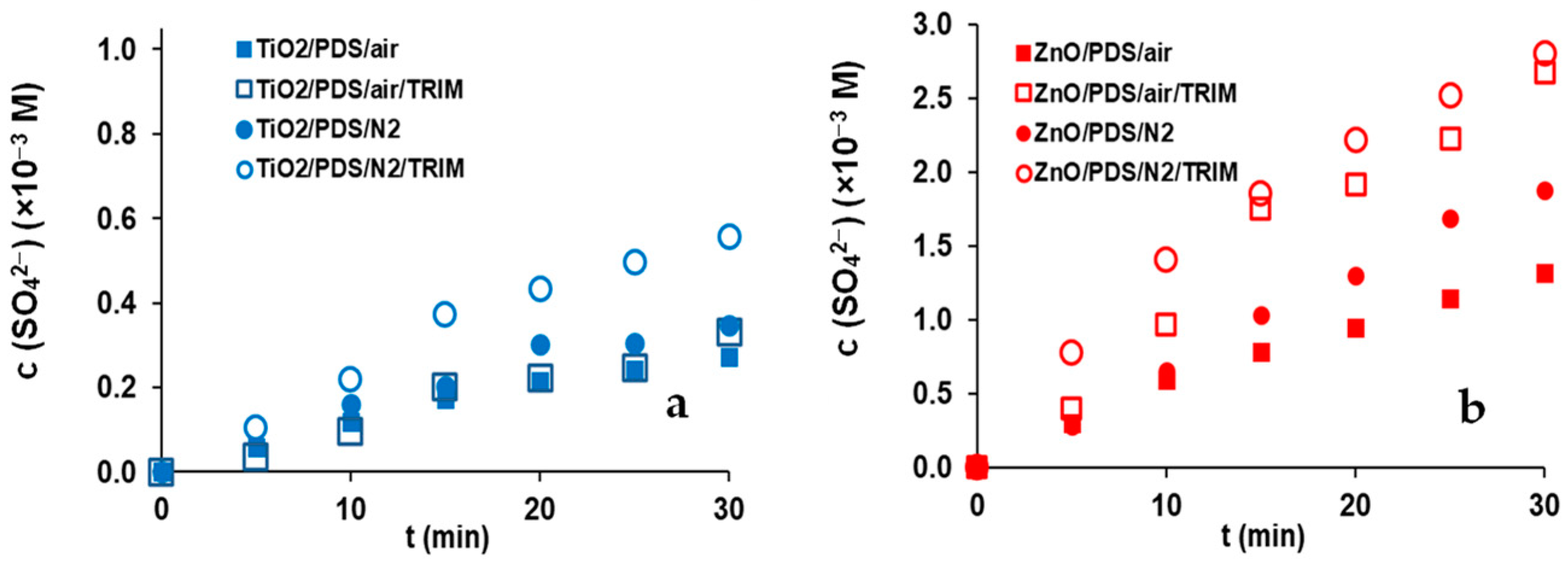
3.3. Effect of Dissolved O2 and Trimethoprim on PDS Transformation
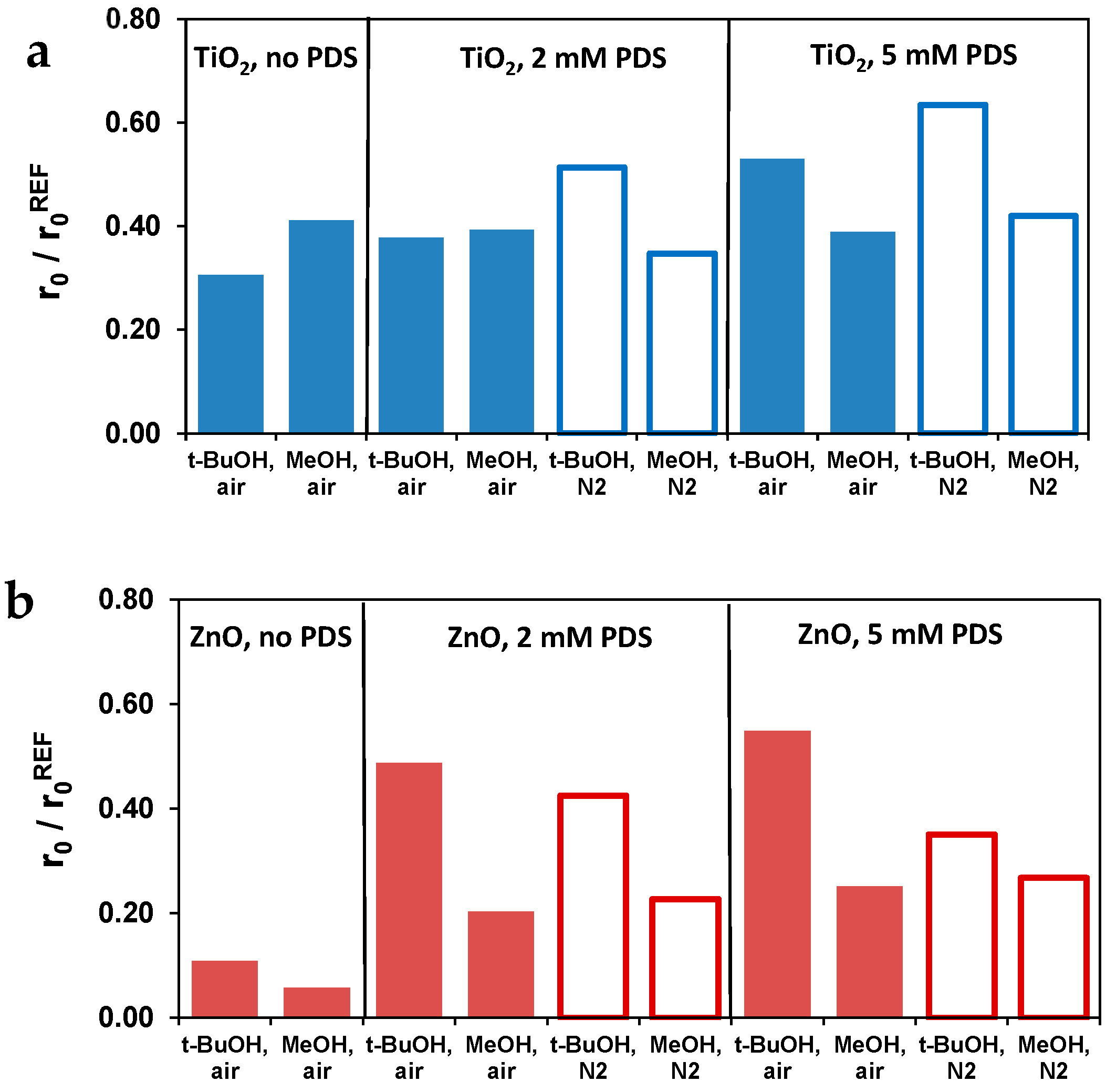
3.4. Effect of Radical Scavengers
3.5. Aromatic Intermediates of TRIM Transformation
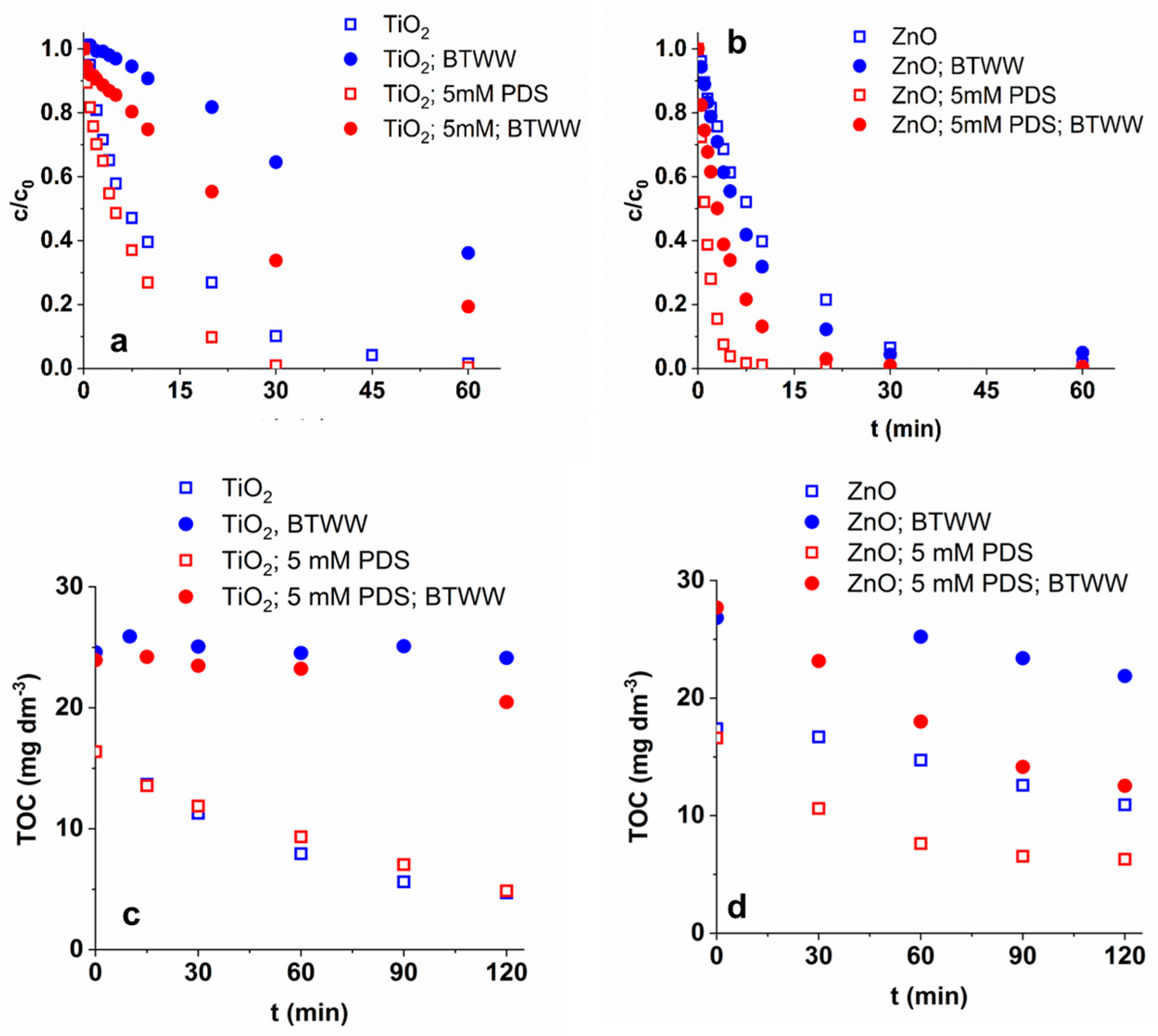
3.6. Matrix Effect
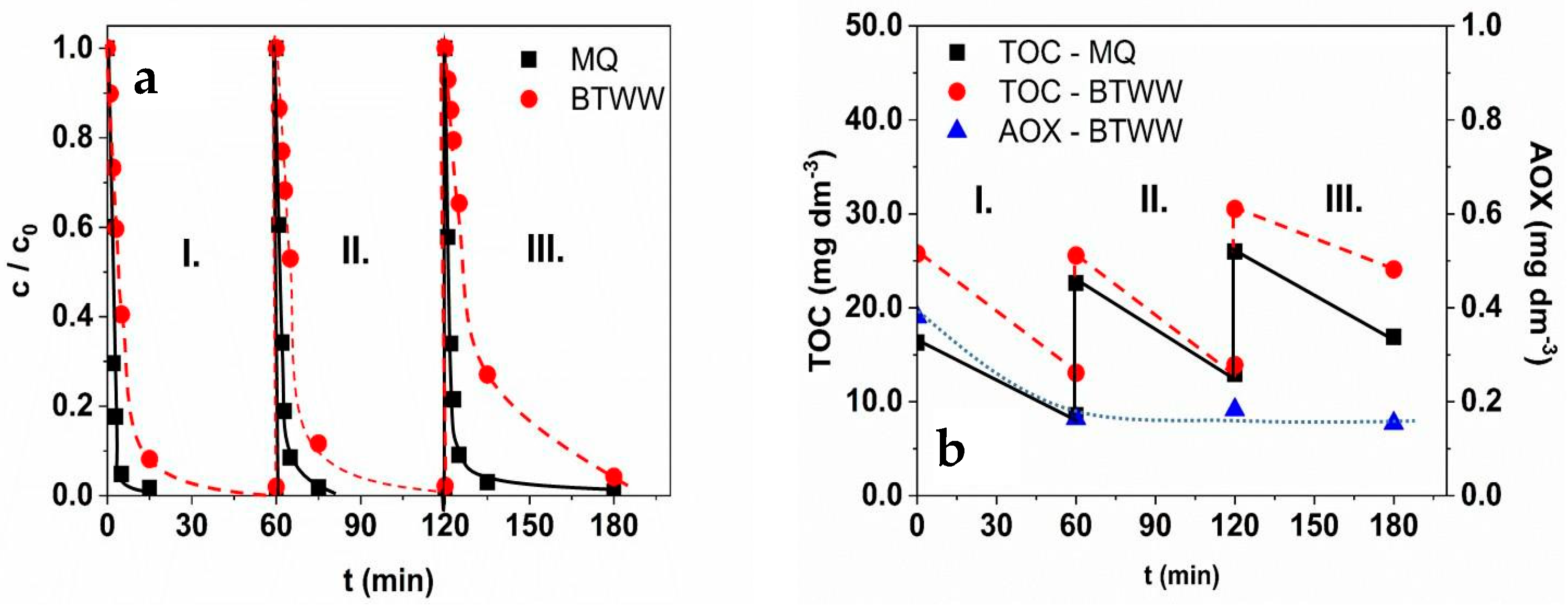
3.7. Reusability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uddin, T.M.; Chakraborty, A.J.; Khusro, A.; Zidan, B.R.M.; Mitra, S.; Emran, T.B.; Dhama, K.; Ripon, M.K.H.; Gajdács, M.; Sahibzada, M.U.K.; et al. Antibiotic Resistance in Microbes: History, Mechanisms, Therapeutic Strategies and Future Prospects. J. Infect. Public. Health 2021, 14, 1750–1766. [Google Scholar] [CrossRef] [PubMed]
- Romandini, A.; Pani, A.; Schenardi, P.A.; Pattarino, G.A.C.; De Giacomo, C.; Scaglione, F. Antibiotic Resistance in Pediatric Infections: Global Emerging Threats, Predicting the near Future. Antibiotics 2021, 10, 393. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Kurittu, P.; Al-Mustapha, A.I.; Heljanko, V.; Johansson, V.; Thakali, O.; Mishra, S.K.; Lehto, K.M.; Lipponen, A.; Oikarinen, S.; et al. Wastewater Surveillance of Antibiotic-Resistant Bacterial Pathogens: A Systematic Review. Front. Microbiol. 2022, 13, 977106. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Long, X.; Wang, X.; Li, L.; Mao, D.; Luo, Y.; Ren, H. Global Trend of Antimicrobial Resistance in Common Bacterial Pathogens in Response to Antibiotic Consumption. J. Hazard. Mater. 2023, 442, 130042. [Google Scholar] [CrossRef]
- Manaia, C.M.; Rocha, J.; Scaccia, N.; Marano, R.; Radu, E.; Biancullo, F.; Cerqueira, F.; Fortunato, G.; Iakovides, I.C.; Zammit, I.; et al. Antibiotic Resistance in Wastewater Treatment Plants: Tackling the Black Box. Environ. Int. 2018, 115, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Pal, D. Antibiotic Resistance and Wastewater: Correlation, Impact and Critical Human Health Challenges. J. Environ. Chem. Eng. 2018, 6, 52–58. [Google Scholar] [CrossRef]
- Stefan, M.I. Sulfate Radical Ion—Based AOPs. In Advanced Oxidation Processes for Water Treatment: Fundamentals and Applications; IWA Publishing: London, UK, 2017; pp. 429–460. [Google Scholar]
- Lee, J.; Von Gunten, U.; Kim, J.H. Persulfate-Based Advanced Oxidation: Critical Assessment of Opportunities and Roadblocks. Environ. Sci. Technol. 2020, 54, 3064–3081. [Google Scholar] [CrossRef]
- Li, H.; Shang, J.; Yang, Z.; Shen, W.; Ai, Z.; Zhang, L. Oxygen Vacancy Associated Surface Fenton Chemistry: Surface Structure Dependent Hydroxyl Radicals Generation and Substrate Dependent Reactivity. Environ. Sci. Technol. 2017, 51, 5685–5694. [Google Scholar] [CrossRef]
- Oh, W.D.; Dong, Z.; Lim, T.T. Generation of Sulfate Radical through Heterogeneous Catalysis for Organic Contaminants Removal: Current Development, Challenges and Prospects. Appl. Catal. B 2016, 194, 169–201. [Google Scholar] [CrossRef]
- Hasija, V.; Nguyen, V.H.; Kumar, A.; Raizada, P.; Krishnan, V.; Khan, A.A.P.; Singh, P.; Lichtfouse, E.; Wang, C.; Thi Huong, P. Advanced Activation of Persulfate by Polymeric G-C3N4 Based Photocatalysts for Environmental Remediation: A Review. J. Hazard. Mater. 2021, 413, 125324. [Google Scholar] [CrossRef]
- Brienza, M.; Katsoyiannis, I.A. Sulfate Radical Technologies as Tertiary Treatment for the Removal of Emerging Contaminants from Wastewater. Sustainability 2017, 9, 1604. [Google Scholar] [CrossRef]
- He, S.; Chen, Y.; Li, X.; Zeng, L.; Zhu, M. Heterogeneous Photocatalytic Activation of Persulfate for the Removal of Organic Contaminants in Water: A Critical Review. ACS ES T Eng. 2022, 2, 527–546. [Google Scholar] [CrossRef]
- Gao, Y.Q.; Gao, N.Y.; Deng, Y.; Yang, Y.Q.; Ma, Y. Ultraviolet (UV) Light-Activated Persulfate Oxidation of Sulfamethazine in Water. Chem. Eng. J. 2012, 195–196, 248–253. [Google Scholar] [CrossRef]
- Saien, J.; Osali, M.; Soleymani, A.R. UV/Persulfate and UV/Hydrogen Peroxide Processes for the Treatment of Salicylic Acid: Effect of Operating Parameters, Kinetic, and Energy Consumption. Desalination Water Treat. 2015, 56, 3087–3095. [Google Scholar] [CrossRef]
- Ji, Y.; Xie, W.; Fan, Y.; Shi, Y.; Kong, D.; Lu, J. Degradation of Trimethoprim by Thermo-Activated Persulfate Oxidation: Reaction Kinetics and Transformation Mechanisms. Chem. Eng. J. 2016, 286, 16–24. [Google Scholar] [CrossRef]
- Ngo Thi, T.D.; Nguyen, L.H.; Nguyen, X.H.; Phung, H.V.; The Vinh, T.H.; Van Viet, P.; Van Thai, N.; Le, H.N.; Pham, D.T.; Van, H.T.; et al. Enhanced Heterogeneous Photocatalytic Perozone Degradation of Amoxicillin by ZnO Modified TiO2 Nanocomposites under Visible Light Irradiation. Mater. Sci. Semicond. Process 2022, 142, 106456. [Google Scholar] [CrossRef]
- Ma, J.; Zhou, H.; Yan, S.; Song, W. Kinetics Studies and Mechanistic Considerations on the Reactions of Superoxide Radical Ions with Dissolved Organic Matter. Water Res. 2019, 149, 56–64. [Google Scholar] [CrossRef]
- Nosaka, Y.; Nosaka, A.Y. Generation and Detection of Reactive Oxygen Species in Photocatalysis. Chem. Rev. 2017, 117, 11302–11336. [Google Scholar] [CrossRef]
- Ge, M.; Hu, Z.; Wei, J.; He, Q.; He, Z. Recent Advances in Persulfate-Assisted TiO2-Based Photocatalysis for Wastewater Treatment: Performances, Mechanism and Perspectives. J. Alloys Compd. 2021, 888, 161625. [Google Scholar] [CrossRef]
- Alapi, T.; Veres, B.; Náfrádi, M.; Farkas, L.; Pap, Z.; Covic, A. Application of BiOX Photocatalyst to Activate Peroxydisulfate Ion-Investigation of a Combined Process for the Removal of Organic Pollutants from Water. Catalysts 2023, 13, 513. [Google Scholar] [CrossRef]
- Oh, W.D.; Lim, T.T. Design and Application of Heterogeneous Catalysts as Peroxydisulfate Activator for Organics Removal: An Overview. Chem. Eng. J. 2019, 358, 110–133. [Google Scholar] [CrossRef]
- Tian, D.; Zhou, H.; Zhang, H.; Zhou, P.; You, J.; Yao, G.; Pan, Z.; Liu, Y.; Lai, B. Heterogeneous Photocatalyst-Driven Persulfate Activation Process under Visible Light Irradiation: From Basic Catalyst Design Principles to Novel Enhancement Strategies. Chem. Eng. J. 2022, 428, 131166. [Google Scholar] [CrossRef]
- Chen, G.; Yu, Y.; Liang, L.; Duan, X.; Li, R.; Lu, X.; Yan, B.; Li, N.; Wang, S. Remediation of Antibiotic Wastewater by Coupled Photocatalytic and Persulfate Oxidation System: A Critical Review. J. Hazard. Mater. 2021, 408, 124461. [Google Scholar] [CrossRef] [PubMed]
- Sabri, M.; Habibi-Yangjeh, A.; Khataee, A. Nanoarchitecturing TiO2/NiCr2O4 p-n Heterojunction Photocatalysts for Visible-Light-Induced Activation of Persulfate to Remove Tetracycline Hydrochloride. Chemosphere 2022, 300, 134594. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhu, M.; Dionysiou, D.D. What Is the Role of Light in Persulfate-Based Advanced Oxidation for Water Treatment? Water Res. 2021, 189, 116627. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Fu, Y.; Li, X.; Wang, L.; Hou, M.; Hu, D.; Li, Q.; Zhang, Z.; Xu, C.; Qiu, S.; et al. Application of Persulfate-Based Oxidation Processes to Address Diverse Sustainability Challenges: A Critical Review. J. Hazard. Mater. 2022, 440, 129722. [Google Scholar] [CrossRef]
- Hatchard, C.G.; Parker, C.A. A New Sensitive Chemical Actinometer II. Potassium Ferrioxalate as a Standard Chemical Actinometer. Proc. R. Soc. Lond. Ser. A 1956, A235, 518–536. [Google Scholar] [CrossRef]
- Zuccheri, T.; Colonna, M.; Stefanini, I.; Santini, C.; Di Gioia, D. Bactericidal Activity of Aqueous Acrylic Paint Dispersion for Wooden Substrates Based on TiO2 Nanoparticles Activated by Fluorescent Light. Materials 2013, 6, 3270–3283. [Google Scholar] [CrossRef]
- White, L.; Koo, Y.; Yun, Y.; Sankar, J. TiO2 Deposition on AZ31 Magnesium Alloy Using Plasma Electrolytic Oxidation. J. Nanomater. 2013, 2013, 319437. [Google Scholar] [CrossRef]
- Thamaphat, K.; Limsuwan, P.; Ngotawornchai, B. Phase Characterization of TiO2 Powder by XRD and TEM. Agric. Nat. Resour. 2008, 42, 357–361. [Google Scholar]
- Arefi, M.R.; Rezaei-Zarchi, S. Synthesis of Zinc Oxide Nanoparticles and Their Effect on the Compressive Strength and Setting Time of Self-Compacted Concrete Paste as Cementitious Composites. Int. J. Mol. Sci. 2012, 13, 4340–4350. [Google Scholar] [CrossRef]
- Náfrádi, M.; Alapi, T.; Bencsik, G.; Janáky, C. Impact of Reaction Parameters and Water Matrices on the Removal of Organic Pollutants by TiO2/LED and ZnO/LED Heterogeneous Photocatalysis Using 365 and 398 nm Radiation. Nanomaterials 2022, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Zheng, Z.; Greaves, J.; Cooper, W.J.; Song, W. Trimethoprim: Kinetic and Mechanistic Considerations in Photochemical Environmental Fate and AOP Treatment. Water Res. 2012, 46, 1327–1336. [Google Scholar] [CrossRef]
- Luo, Y.; Su, R.; Yao, H.; Zhang, A.; Xiang, S.; Huang, L. Degradation of Trimethoprim by Sulfate Radical-Based Advanced Oxidation Processes: Kinetics, Mechanisms, and Effects of Natural Water Matrices. Environ. Sci. Pollut. Res. 2021, 28, 62572–62582. [Google Scholar] [CrossRef]
- Zeng, M. Influence of TiO2 Surface Properties on Water Pollution Treatment and Photocatalytic Activity. Bull. Korean Chem. Soc. 2013, 34, 953–956. [Google Scholar] [CrossRef]
- Kosmulski, M. The Significance of the Difference in the Point of Zero Charge between Rutile and Anatase. Adv. Colloid. Interface Sci. 2002, 99, 255–264. [Google Scholar] [CrossRef]
- Degen, A.; Kosec, M. Effect of pH and Impurities on the Surface Charge of Zinc Oxide in Aqueous Solution. J. Eur. Ceram. Soc. 2000, 20, 667–673. [Google Scholar] [CrossRef]
- Le, A.T.; Syuhada, N.; Samsuddin, B.; Chiam, S.-L.; Pung, S.-Y. Synergistic Effect of pH Solution and Photocorrosion of ZnO Particles on the Photocatalytic Degradation of Rhodamine B. Bull. Mater. Sci. 2021, 44, 5. [Google Scholar] [CrossRef]
- Taylor, Z.; Marucho, M. The Self-Adaptation Ability of Zinc Oxide Nanoparticles Enables Reliable Cancer Treatments. Nanomaterials 2020, 10, 269. [Google Scholar] [CrossRef]
- Fatehah, M.O.; Aziz, H.A.; Stroll, S. Stability of ZnO Nanoparticles in Solution. Influence of pH, Dissolution, Aggregation and Disaggregation Effects. J. Colloid. Sci. Biotechnol. 2014, 3, 75–84. [Google Scholar] [CrossRef]
- Rao, S.R.; Finch, J.A. Base Metal Oxide Flotation Using Long Chain Xanthates. Int. J. Miner. Process. 2003, 69, 251–258. [Google Scholar] [CrossRef]
- Do, T.H.; Nguyen, V.T.; Nguyen, Q.D.; Chu, M.N.; Ngo, T.C.Q.; Van Tan, L. Equilibrium, Kinetic and Thermodynamic Studies for Sorption of Phosphate from Aqueous Solutions Using Zno Nanoparticles. Processes 2020, 8, 1397. [Google Scholar] [CrossRef]
- Liu, C.-F.; Lu, Y.-J.; Hu, C.-C. Effects of Anions and PH on the Stability of ZnO Nanorods for Photoelectrochemical Water Splitting. ACS Omega 2018, 3, 3429–3439. [Google Scholar] [CrossRef]
- Janusz, W. Specific Adsorption of Carbonate Ions at the Zinc Oxide/Electrolyte Solution Interface Article in Physicochemical Problems of Mineral Processing. Physicochem. Probl. Miner. Process. 2008, 42, 57–66. [Google Scholar]
- Chen, Q.; Wang, H.; Luan, Q.; Duan, R.; Cao, X.; Fang, Y.; Ma, D.; Guan, R.; Hu, X. Synergetic Effects of Defects and Acid Sites of 2D-ZnO Photocatalysts on the Photocatalytic Performance. J. Hazard. Mater. 2020, 385, 121527. [Google Scholar] [CrossRef] [PubMed]
- Baxter, J.B.; Schmuttenmaer, C.A. Conductivity of ZnO Nanowires, Nanoparticles, and Thin Films Using Time-Resolved Terahertz Spectroscopy. J. Phys. Chem. B 2006, 110, 25229–25239. [Google Scholar] [CrossRef]
- Meulenkamp, E.A. Electron Transport in Nanoparticulate ZnO Films. J. Phys. Chem. B 1999, 103, 7831–7838. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Moon, G.h.; Kim, B.; Tachikawa, T.; Majima, T.; Hong, S.; Cho, K.; Kim, W.; Choi, W. Crystal Phase-Dependent Generation of Mobile OH Radicals on TiO2: Revisiting the Photocatalytic Oxidation Mechanism of Anatase and Rutile. Appl. Catal. B 2021, 286, 119905. [Google Scholar] [CrossRef]
- Neta, P.; Madhavan, V.; Zemel, H.; Fessenden, R.W. Rate Constants and Mechanism of Reaction of Sulfate Radical Anion with Aromatic Compounds. J. Am. Chem. Soc. 1977, 99, 163–164. [Google Scholar] [CrossRef]
- Yu, X.-Y.; Bao, Z.-C.; Barker, J. Free Radical Reactions Involving Cl•, Cl2−•, and SO4−• in the 248 nm Photolysis of Aqueous Solutions Containing S2O82− and Cl−. J. Phys. Chem. A 2003, 35, 295–308. [Google Scholar] [CrossRef]
- Klaning, U.K.; Sehested, K.; Appelman, E.H. Laser Flash Photolysis and Pulse Radiolysis of Aqueous Solutions of the Fluoroxysulfate Ion, SO4F−. Inorg. Chem. 1991, 30, 3582–3584. [Google Scholar] [CrossRef]
- Jo, Y.; Kim, C.; Moon, G.H.; Lee, J.; An, T.; Choi, W. Activation of Peroxymonosulfate on Visible Light Irradiated TiO2 via a Charge Transfer Complex Path. Chem. Eng. J. 2018, 346, 249–257. [Google Scholar] [CrossRef]
- Chen, X.; Wang, W.; Xiao, H.; Hong, C.; Zhu, F.; Yao, Y.; Xue, Z. Accelerated TiO2 Photocatalytic Degradation of Acid Orange 7 under Visible Light Mediated by Peroxymonosulfate. Chem. Eng. J. 2012, 193–194, 290–295. [Google Scholar] [CrossRef]
- Fang, G.; Gao, J.; Dionysiou, D.D.; Liu, C.; Zhou, D. Activation of Persulfate by Quinones: Free Radical Reactions and Implication for the Degradation of PCBs. Environ. Sci. Technol. 2013, 47, 4605–4611. [Google Scholar] [CrossRef] [PubMed]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals (•OH/•O− in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Hu, L.; Wang, P.; Shen, T.; Wang, Q.; Wang, X.; Xu, P.; Zheng, Q.; Zhang, G. The Application of Microwaves in Sulfate Radical-Based Advanced Oxidation Processes for Environmental Remediation: A Review. Sci. Total Environ. 2020, 722, 137831. [Google Scholar] [CrossRef]
- Heckel, E. Pulsradiolytische Untersuchung Des Radikal-Anions SO42−. Angew. Chem. 1966, 78, 779. [Google Scholar] [CrossRef]
- Samy, M.; Ibrahim, M.G.; Gar Alalm, M.; Fujii, M.; Ookawara, S.; Ohno, T. Photocatalytic Degradation of Trimethoprim Using S-TiO2 and Ru/WO3/ZrO2 Immobilized on Reusable Fixed Plates. J. Water Process Eng. 2020, 33, 101023. [Google Scholar] [CrossRef]
- Liu, J.; Ding, C.; Gong, S.; Fu, K.; Deng, H.; Shi, J. Enhanced Degradation of Antibiotic by Peroxydisulfate Catalysis with CuO@CNT: Simultaneous 1O2 Oxidation and Electron-Transfer Regime. Molecules 2022, 27, 7064. [Google Scholar] [CrossRef]
- Scaria, J.; Nidheesh, P.V. Comparison of Hydroxyl-Radical-Based Advanced Oxidation Processes with Sulfate Radical-Based Advanced Oxidation Processes. Curr. Opin. Chem. Eng. 2022, 36, 100830. [Google Scholar] [CrossRef]
- Xia, X.; Zhu, F.; Li, J.; Yang, H.; Wei, L.; Li, Q.; Jiang, J.; Zhang, G.; Zhao, Q. A Review Study on Sulfate-Radical-Based Advanced Oxidation Processes for Domestic/Industrial Wastewater Treatment: Degradation, Efficiency, and Mechanism. Front. Chem. 2020, 8, 592056. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.; Agüera, A.; Gernjak, W.; Malato, S. Effect of Water-Matrix Composition on Trimethoprim Solar Photodegradation Kinetics and Pathways. Water Res. 2010, 44, 2735–2744. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Hu, J. Decomposition of Sulfamethoxazole and Trimethoprim by Continuous UVA/LED/TiO2 Photocatalysis: Decomposition Pathways, Residual Antibacterial Activity and Toxicity. J. Hazard. Mater. 2017, 323, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, Q.; Wang, J. Degradation of Trimethoprim by Gamma Irradiation in the Presence of Persulfate. Radiat. Phys. Chem. 2016, 127, 85–91. [Google Scholar] [CrossRef]
- Farner Budarz, J.; Turolla, A.; Piasecki, A.F.; Bottero, J.Y.; Antonelli, M.; Wiesner, M.R. Influence of Aqueous Inorganic Anions on the Reactivity of Nanoparticles in TiO2 Photocatalysis. Langmuir 2017, 33, 2770–2779. [Google Scholar] [CrossRef]
- Kaabeche, O.N.E.H.; Zouaghi, R.; Boukhedoua, S.; Bendjabeur, S.; Sehili, T. A Comparative Study on Photocatalytic Degradation of Pyridinium-Based Ionic Liquid by TiO2 and ZnO in Aqueous Solution. Int. J. Chem. React. Eng. 2019, 17, 20180253. [Google Scholar] [CrossRef]
- Lair, A.; Ferronato, C.; Chovelon, J.M.; Herrmann, J.M. Naphthalene Degradation in Water by Heterogeneous Photocatalysis: An Investigation of the Influence of Inorganic Anions. J. Photochem. Photobiol. A Chem. 2008, 193, 193–203. [Google Scholar] [CrossRef]
- Šojić Merkulov, D.V.; Lazarević, M.J.; Despotović, V.N.; Banić, N.D.; Finčur, N.L.; Maletić, S.P.; Abramović, B.F. The Effects of Inorganic Anions and Organic Matter on Mesotrione (Callisto®) Removal from Environmental Waters. J. Serbian Chem. Soc. 2017, 82, 343–355. [Google Scholar] [CrossRef]
- Bouanimba, N.; Laid, N.; Zouaghi, R.; Sehili, T. A Comparative Study of the Activity of TiO2 Degussa P25 and Millennium PCs in the Photocatalytic Degradation of Bromothymol Blue. Int. J. Chem. React. Eng. 2018, 16, 20170014. [Google Scholar] [CrossRef]
- Cheng, Z.; Kok Eng, T.; Tao, Y.; Eng Ting, K.; Jiang Yin, X. Studies of Photocatalytic Kinetics on the Degradation of Bisphenol a (BPA) by Immobilized ZnO Nanoparticles in Aerated Photoreactors. J. Environ. Sci. Eng. A 2012, 1, 187–194. [Google Scholar]
- Wojnárovits, L.; Tóth, T.; Takács, E. Rate Constants of Carbonate Radical Anion Reactions with Molecules of Environmental Interest in Aqueous Solution: A Review. Sci. Total Environ. 2020, 717, 137219. [Google Scholar] [CrossRef]
- Stefan, M.I. UV/Chlorine Process. In Advanced Oxidation Processes for Water Treatment: Fundamentals and Applications; IWA Publishing: London, UK, 2017; pp. 383–428. [Google Scholar]
- Khan, S.; He, X.; Khan, J.A.; Khan, H.M.; Boccelli, D.L.; Dionysiou, D.D. Kinetics and Mechanism of Sulfate Radical- and Hydroxyl Radical-Induced Degradation of Highly Chlorinated Pesticide Lindane in UV/Peroxymonosulfate System. Chem. Eng. J. 2017, 318, 135–142. [Google Scholar] [CrossRef]
- Mertens, R.; yon Sonntag, C. Photolysis (λ = 254 nm) of Tetrachloroethene in Aqueous Solutions. J. Photochem. Photobiol. A Chem. 1995, 85, 1–9. [Google Scholar] [CrossRef]
- Dell’Arciprete, M.L.; Soler, J.M.; Santos-Juanes, L.; Arques, A.; Mártire, D.O.; Furlong, J.P.; Gonzalez, M.C. Reactivity of Neonicotinoid Insecticides with Carbonate Radicals. Water Res. 2012, 46, 3479–3489. [Google Scholar] [CrossRef] [PubMed]
- Mazellier, P.; Busset, C.; Delmont, A.; De Laat, J. A Comparison of Fenuron Degradation by Hydroxyl and Carbonate Radicals in Aqueous Solution. Water Res. 2007, 41, 4585–4594. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | TRIM | P1 | P2a | P2b | P3 |
|---|---|---|---|---|---|
| Structure | 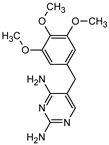 | 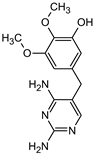 |  | 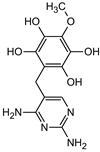 | 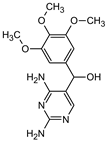 |
| tret (min) | 9.5 | 2.5 | 3.1 | 3.1 | 3.5 |
| m/z (AMU) | 291.2 | 277.2 | 309.2 | 295.2 | 307.2 |
| Name | P4 | P5 | P6 | P7 | P8 |
| Structure | 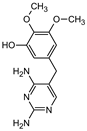 |  | 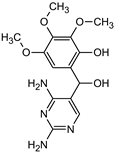 | 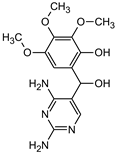 | |
| tret (min) | 3.7 | 5.1 | 5.6 | 6.9 | 7.3 |
| m/z (AMU) | 277.2 | only DAD detection | 325.3 | 323.2 | 307.2 |
| r0 (×10−7 M s−1) | ||||
|---|---|---|---|---|
| TiO2 | TiO2/PDS | ZnO | ZnO/PDS | |
| MQ | 1.72 | 1.95 | 1.58 | 6.83 |
| Cl− | 1.43 | 1.40 | 1.56 | 5.18 |
| HCO3− | 1.03 | 1.27 | 1.39 | 4.08 |
| Cl− + HCO3− | 1.05 | 1.25 | 1.45 | 4.72 |
| BTWW | 0.16 | 0.40 | 1.70 | 3.33 |
| r0BTWW/r0MQ | ||||
| 0.06 | 0.21 | 1.08 | 0.49 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Náfrádi, M.; Alapi, T.; Veres, B.; Farkas, L.; Bencsik, G.; Janáky, C. Comparison of TiO2 and ZnO for Heterogeneous Photocatalytic Activation of the Peroxydisulfate Ion in Trimethoprim Degradation. Materials 2023, 16, 5920. https://doi.org/10.3390/ma16175920
Náfrádi M, Alapi T, Veres B, Farkas L, Bencsik G, Janáky C. Comparison of TiO2 and ZnO for Heterogeneous Photocatalytic Activation of the Peroxydisulfate Ion in Trimethoprim Degradation. Materials. 2023; 16(17):5920. https://doi.org/10.3390/ma16175920
Chicago/Turabian StyleNáfrádi, Máté, Tünde Alapi, Bence Veres, Luca Farkas, Gábor Bencsik, and Csaba Janáky. 2023. "Comparison of TiO2 and ZnO for Heterogeneous Photocatalytic Activation of the Peroxydisulfate Ion in Trimethoprim Degradation" Materials 16, no. 17: 5920. https://doi.org/10.3390/ma16175920