
Enhancing Starch−Based Packaging Materials: Optimization of Plasticizers and Process Parameters

 ,
,  ,
,
Abstract
:
1. Introduction
2. Materials and Experimental Procedures
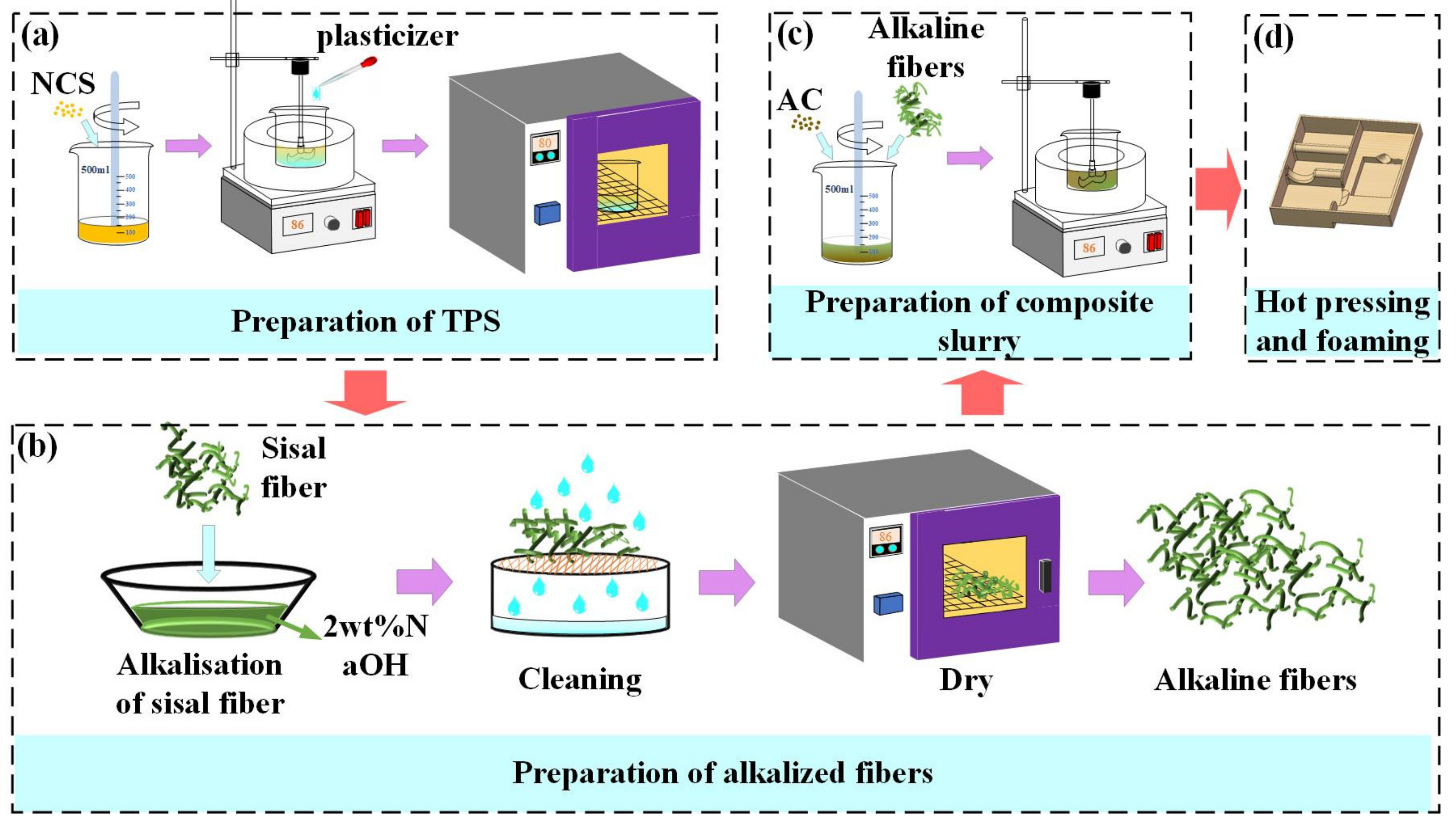
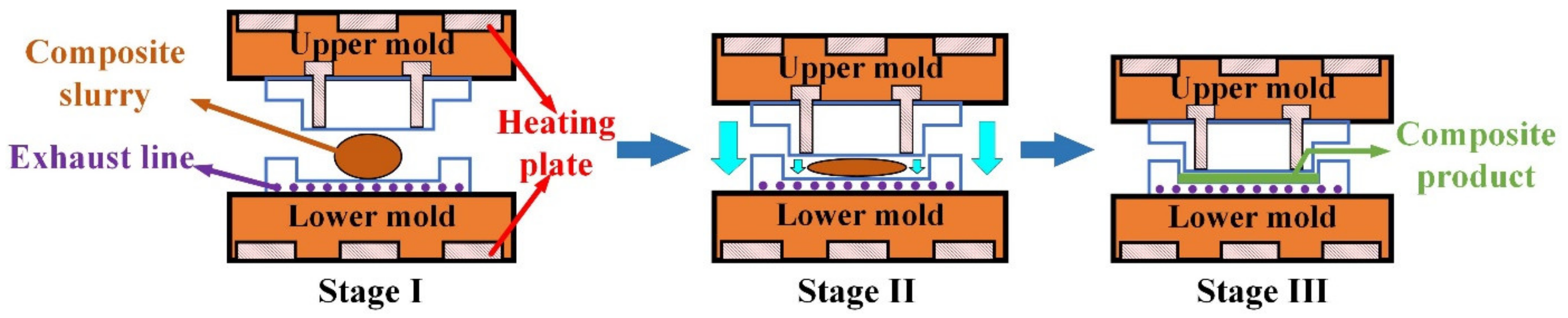
2.1. Materials Preparation and Experimental Set−Up
2.2. Material Characterizations
2.3. Mechanical Performance Testing
2.4. Process Parameter Optimization
- (1)
- Establishing a general mathematical relationship between the input variables and response, which can be expressed as follows:where is the response, represents the mathematical relationship, and , , … …, are input variables.
- (2)
- Looking for mathematical models. Based on mathematical analysis, the existing McLaughlin or Taylor expansions can meet convergence conditions. According to Samir Charola et al. [36], the experimental data can be fitted into a second−order polynomial model, namely:where , , , and are the regression coefficients of the intercept, the linearity, the interaction, and the quadratic terms, respectively. and denote the input variables, and k represents the number of input variables selected for process optimization.
- (3)
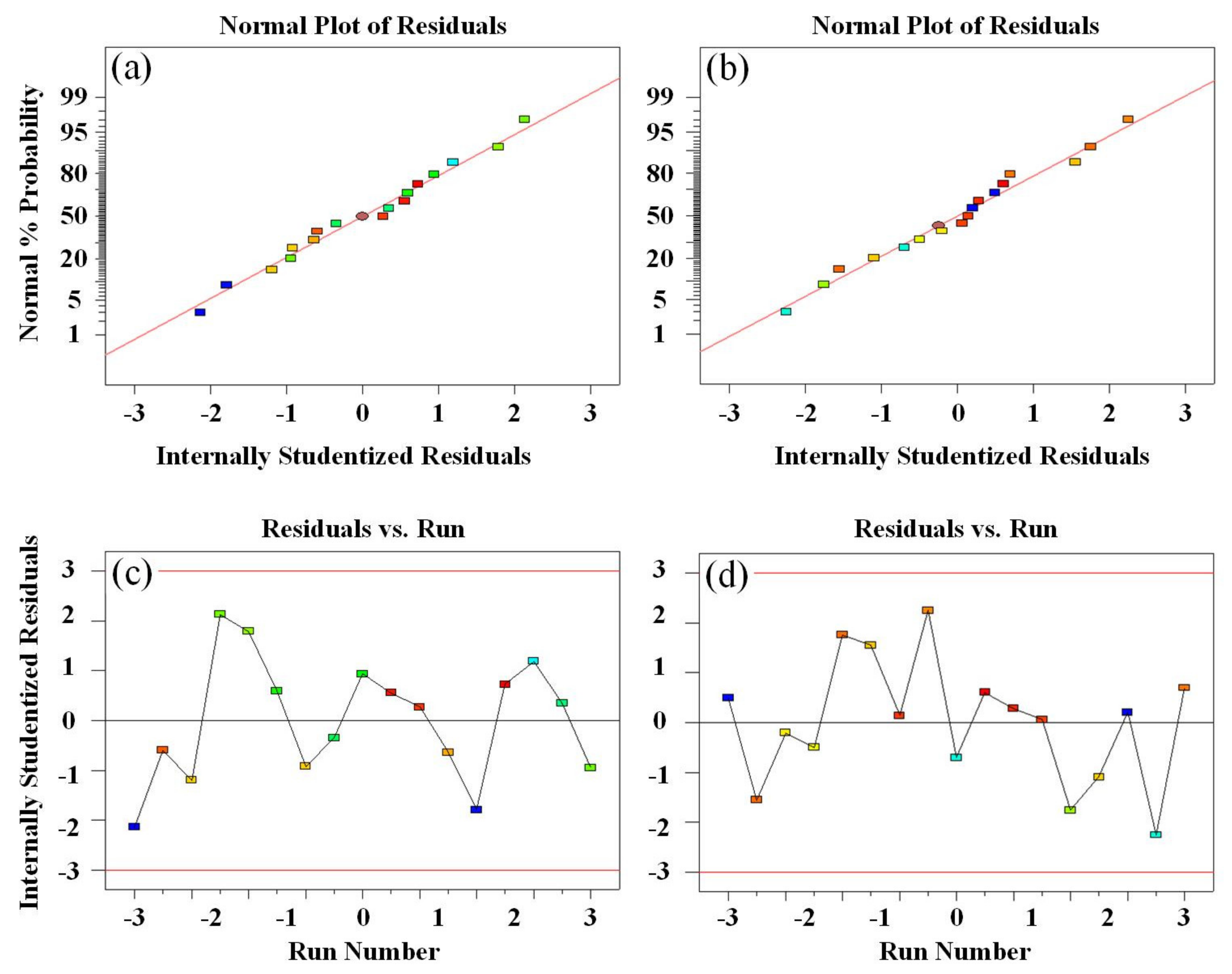
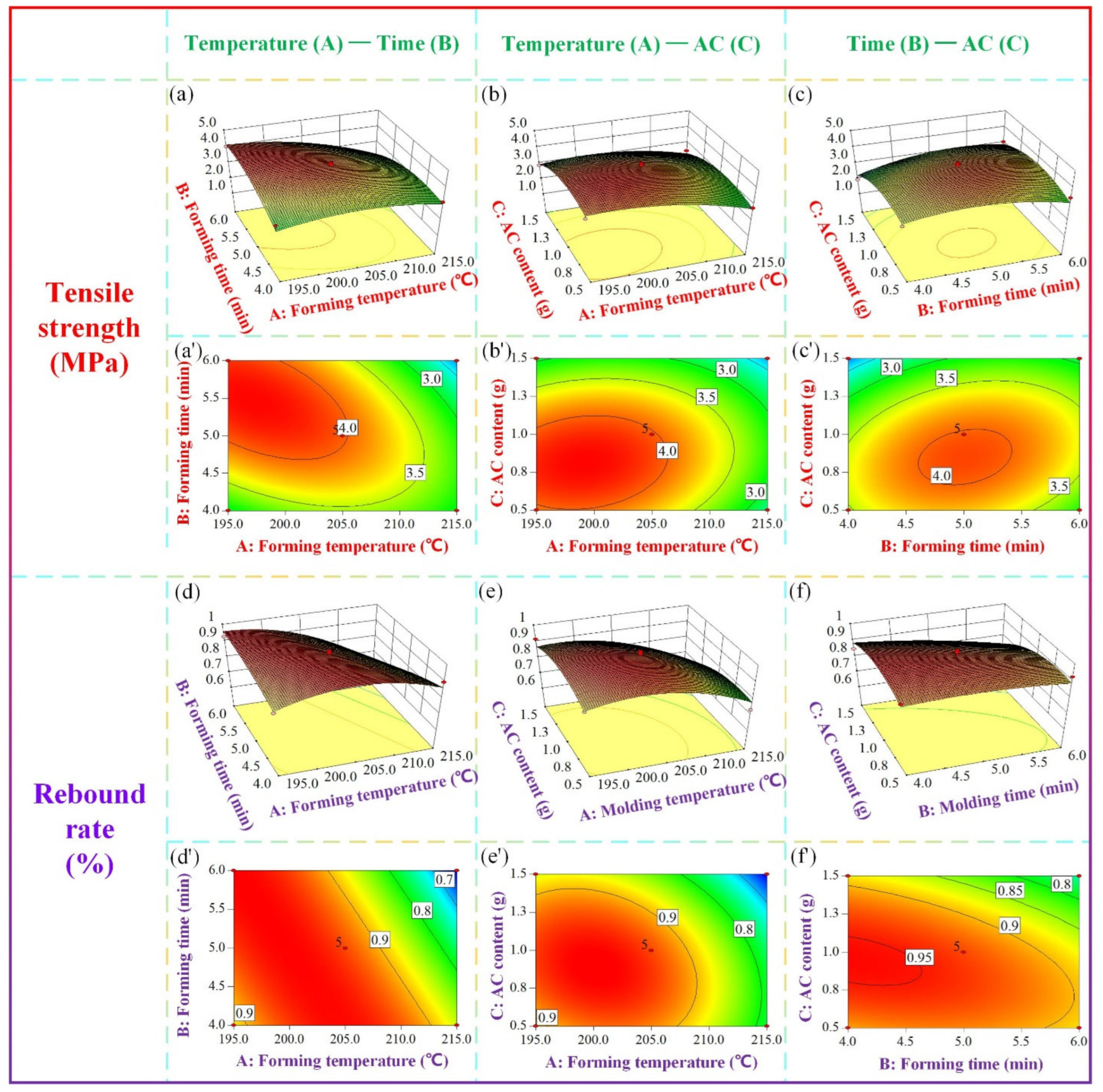
- Using analysis of variance (ANOVA) to evaluate the significance of the prediction model. The reliability of the model can be determined by loss of fit (LOF) and p value. The actual values are the tensile strength and rebound rate measured by experiments. Based on the second−order polynomial model, the response surface and the contour plot, which describe the influences of two variables on a single target, can be obtained. The third input variable remains unchanged at the center point. Then, the effects of input variables on the response will be analyzed to obtain the highest rebound rate and tensile strength simultaneously.
3. Results and Discussion
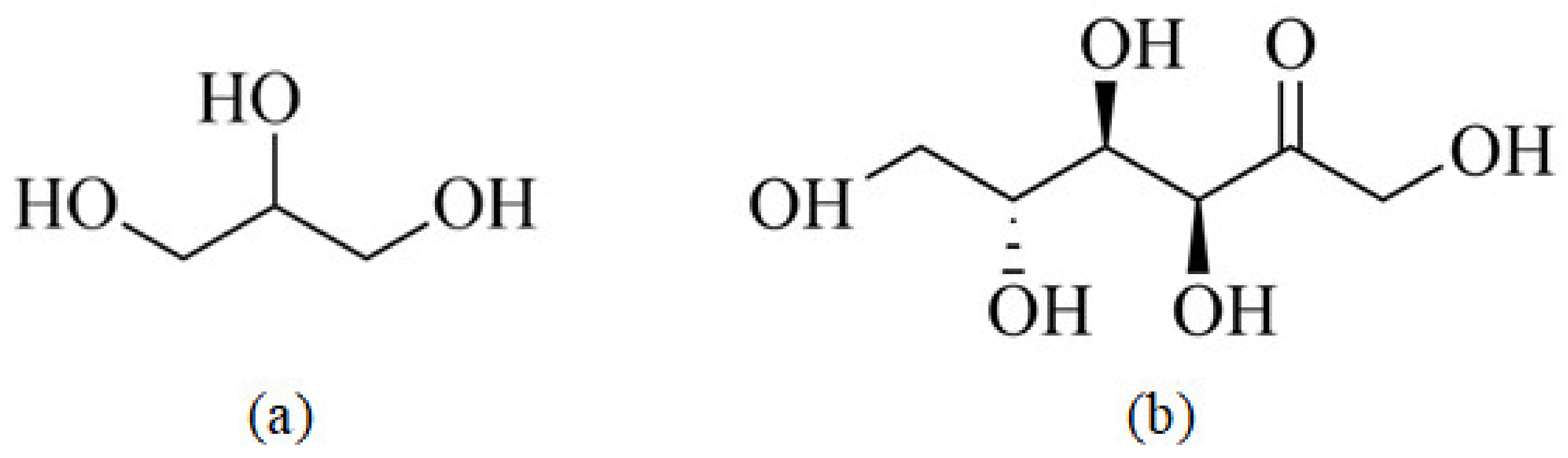
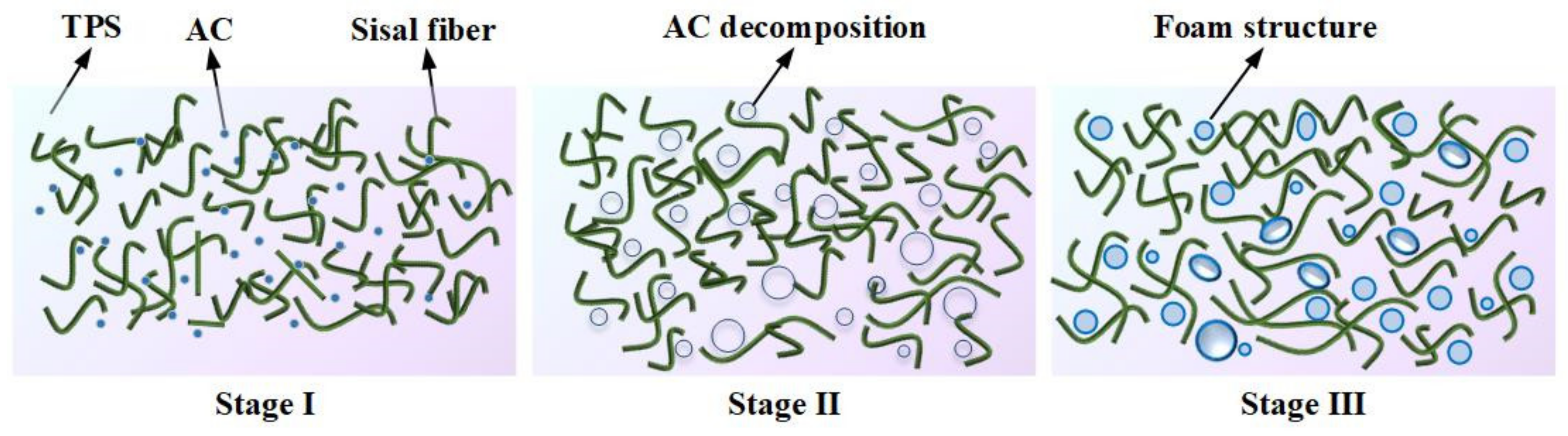
3.1. The Plasticization Process of Starch
3.2. Effects of Plasticizer on TPS
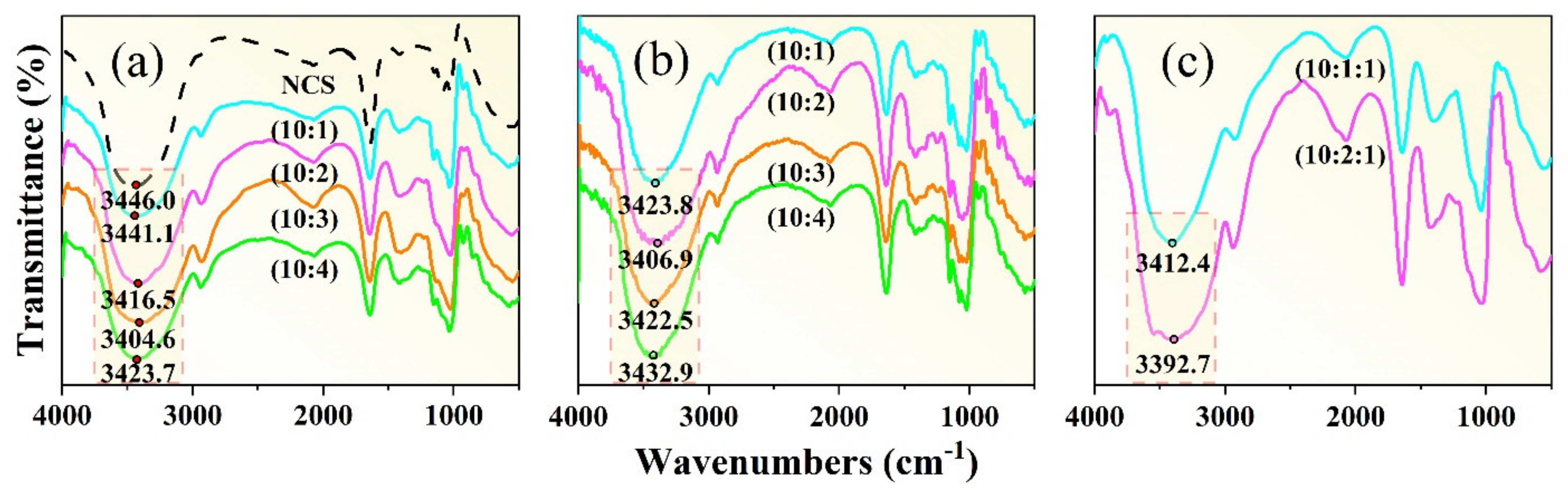
3.2.1. Effects of Plasticizer on Hydrogen Bond
3.2.2. Effects of Plasticizer on Crystallization Type
3.2.3. Effects of Plasticizer on Micromorphology
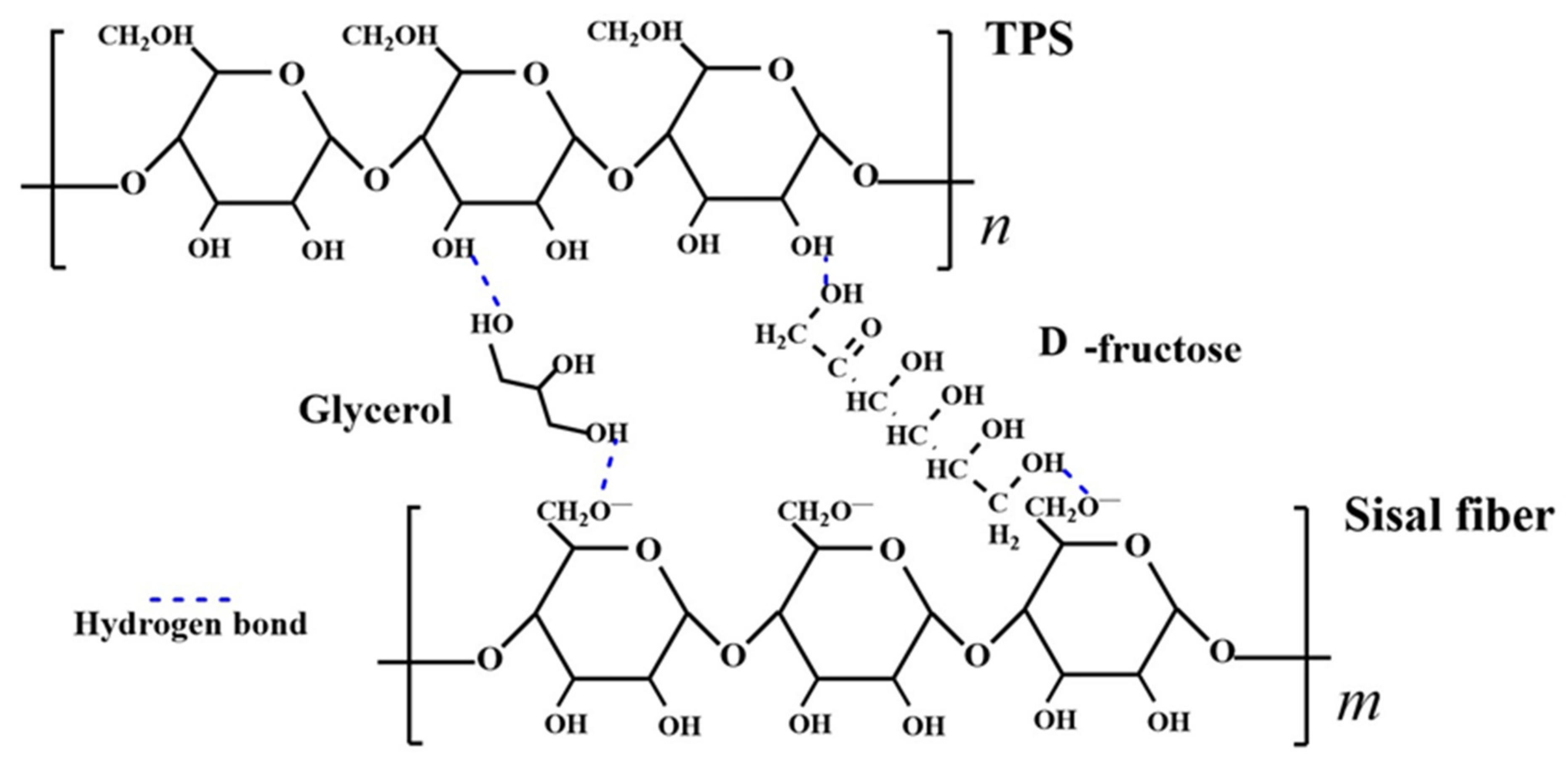
3.3. Molecular Linking Model of Starch−Based Composites
3.4. Parameter Optimization of the Starch Composites
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adeniran, A.A.; Shakantu, W. The Health and Environmental Impact of Plastic Waste Disposal in South African Townships: A Review. Int. J. Environ. Res. Public Health 2022, 19, 779. [Google Scholar] [CrossRef]
- Gaston, S.A.; Tulve, N.S. Urinary phthalate metabolites and metabolic syndrome in U.S. adolescents: Cross-sectional results from the National Health and Nutrition Examination Survey (2003–2014) data. Int. J. Hyg. Environ. Health 2019, 222, 195–204. [Google Scholar] [CrossRef]
- Hahladakis, J.N.; Velis, C.A.; Weber, R.; Iacovidou, E.; Purnell, P. An overview of chemical additives present in plastics: Migration, release, fate and environmental impact during their use, disposal and recycling. J. Hazard. Mater. 2018, 344, 179–199. [Google Scholar] [CrossRef]
- Xu, R.; Chen, F.; Ding, C. Opportunities, challenges and suggestions for the development of plastic degradation and recycling under the context of circular bioeconomy. Sheng Wu Gong Cheng Xue Bao = Chin. J. Biotechnol. 2023, 39, 1867–1882. [Google Scholar] [CrossRef]
- Filho, W.L.; Salvia, A.L.; Bonoli, A.; Saari, U.A.; Voronova, V.; Klõga, M.; Kumbhar, S.S.; Olszewski, K.; De Quevedo, D.M.; Barbir, J. An assessment of attitudes towards plastics and bioplastics in Europe. Sci. Total Environ. 2021, 755, 142732. [Google Scholar] [CrossRef] [PubMed]
- Rajvanshi, J.; Sogani, M.; Kumar, A.; Arora, S.; Syed, Z.; Sonu, K.; Gupta, N.S.; Kalra, A. Perceiving biobased plastics as an alternative and innovative solution to combat plastic pollution for a circular economy. Sci. Total Environ. 2023, 874, 162441. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.-H.; Singh, S.; Carr Clennon, A.N.; Dien, B.S.; Singh, V. Production of Designer Xylose-Acetic Acid Enriched Hydrolysate from Bioenergy Sorghum, Oilcane, and Energycane Bagasses. Bioresour. Technol. 2023, 380, 129104. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, X.; Ren, H.; Hu, X.; Yang, X.; Liu, H. Co-production of spirosiloxane and biochar adsorbent from wheat straw by a low-cost and environment-friendly method. J. Environ. Manag. 2023, 338, 117851. [Google Scholar] [CrossRef] [PubMed]
- Suvarnna, K.; Shanjitha, S.; Selvasekarapandian, S.; Kirubavathy, S.J. Investigation of solid bio-membrane based on corn biomass as a proton-conducting bio-electrolyte. Bull. Mater. Sci. 2023, 46, 112. [Google Scholar] [CrossRef]
- Guo, A.; Tao, X.; Kong, H.; Zhou, X.; Wang, H.; Li, J.; Li, F.; Hu, Y. Effects of aluminum hydroxide on mechanical, water resistance, and thermal properties of starch-based fiber-reinforced composites with foam structures. J. Mater. Res. Technol. 2023, 23, 1570–1583. [Google Scholar] [CrossRef]
- Bangar, S.P.; Purewal, S.S.; Trif, M.; Maqsood, S.; Kumar, M.; Manjunatha, V.; Rusu, A.V. Functionality and Applicability of Starch-Based Films: An Eco-Friendly Approach. Foods 2021, 10, 2181. [Google Scholar] [CrossRef]
- Do Val Siqueira, L.; Arias, C.I.L.F.; Maniglia, B.C.; Tadini, C.C. Starch-based biodegradable plastics: Methods of production, challenges and future perspectives. Curr. Opin. Food Sci. 2021, 38, 122–130. [Google Scholar] [CrossRef]
- Jane, J.-L.; Robyt, J.F. Structure studies of amylose-V complexes and retro-graded amylose by action of alpha amylases, and a new method for preparing amylodextrins. Carbohydr. Res. 1984, 132, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Biduski, B.; Silva, W.M.F.D.; Colussi, R.; Halal, S.L.D.M.E.; Lim, L.-T.; Dias, Á.R.G.; Zavareze, E.D.R. Starch hydrogels: The influence of the amylose content and gelatinization method. Int. J. Biol. Macromol. 2018, 113, 443–449. [Google Scholar] [CrossRef]
- Zhu, F.; Liu, P. Starch gelatinization, retrogradation, and enzyme susceptibility of retrograded starch: Effect of amylopectin internal molecular structure. Food Chem. 2020, 316, 126036. [Google Scholar] [CrossRef]
- Cheng, W. Preparation and properties of lignocellulosic fiber/CaCO3/thermoplastic starch composites. Carbohydr. Polym. 2019, 211, 204–208. [Google Scholar] [CrossRef]
- Zhang, B.; Xie, F.; Zhang, T.; Chen, L.; Li, X.; Truss, R.W.; Halley, P.J.; Shamshina, J.L.; McNally, T.; Rogers, R.D. Different characteristic effects of ageing on starch-based films plasticised by 1-ethyl-3-methylimidazolium acetate and by glycerol. Carbohydr. Polym. 2016, 146, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Kahvand, F.; Fasihi, M. Plasticizing and anti-plasticizing effects of polyvinyl alcohol in blend with thermoplastic starch. Int. J. Biol. Macromol. 2019, 140, 775–781. [Google Scholar] [CrossRef]
- Ávila-Martín, L.; Beltrán-Osuna, Á.A.; Perilla, J.E. Effect of the Addition of Citric Acid and Whey Protein Isolate in Canna indica L. Starch Films Obtained by Solvent Casting. J. Polym. Environ. 2020, 28, 871–883. [Google Scholar] [CrossRef]
- Zeng, G.-S.; Lin, R.-Z.; Zheng, L.-J.; Chen, L.; Meng, C. Preparation and performance study of waste paper pulp reinforced cornstarch-based composites. J. Funct. Mater. 2012, 43, 2218–2221. [Google Scholar]
- De Graaf, R.A.; Karman, A.P.; Janssen, L.P.B.M. Material Properties and Glass Transition Temperatures of Different Thermoplastic Starches After Extrusion Processing. Starch-Stärke 2003, 55, 80–86. [Google Scholar] [CrossRef]
- Enrione, J.; Osorio, F.; Pedreschi, F.; Hill, S. Prediction of the Glass Transition Temperature on Extruded Waxy Maize and Rice Starches in Presence of Glycerol. Food Bioprocess Technol. 2010, 3, 791–796. [Google Scholar] [CrossRef]
- Schmitt, H.; Guidez, A.; Prashantha, K.; Soulestin, J.; Lacrampe, M.F.; Krawczak, P. Studies on the effect of storage time and plasticizers on the structural variations in thermoplastic starch. Carbohydr. Polym. 2015, 115, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Zdanowicz, M.; Staciwa, P.; Spychaj, T. Low Transition Temperature Mixtures (LTTM) Containing Sugars as Potato Starch Plasticizers. Starch-Stärke 2019, 71, 1900004. [Google Scholar] [CrossRef]
- Saberi, B.; Chockchaisawasdee, S.; Golding, J.B.; Scarlett, C.J.; Stathopoulos, C.E. Physical and mechanical properties of a new edible film made of pea starch and guar gum as affected by glycols, sugars and polyols. Int. J. Biol. Macromol. 2017, 104, 345–359. [Google Scholar] [CrossRef]
- Müller, P.; Renner, K.; Móczó, J.; Fekete, E.; Pukánszky, B. Thermoplastic starch/wood composites: Interfacial interactions and functional properties. Carbohydr. Polym. 2014, 102, 821–829. [Google Scholar] [CrossRef]
- Mohammed, A.A.B.A.; Hasan, Z.; Omran, A.A.B.; Kumar, V.V.; Elfaghi, A.M.; Ilyas, R.A.; Sapuan, S.M. Corn: Its Structure, Polymer, Fiber, Composite, Properties, and Applications. Polymers 2022, 14, 4396. [Google Scholar] [CrossRef]
- Jenkins, P.J.; Donald, A.M. The influence of amylose on starch granule structure. Int. J. Biol. Macromol. 1995, 17, 315–321. [Google Scholar] [CrossRef]
- Zhang, C.-W.; Li, F.-Y.; Li, J.-F.; Xie, Q.; Xu, J.; Guo, A.-F.; Wang, C.-Z. Effect of Crystal Structure and Hydrogen Bond of Thermoplastic Oxidized Starch on Manufacturing of Starch-Based Biomass Composite. Int. J. Precis. Eng. Manuf.-Green Technol. 2018, 5, 435–440. [Google Scholar] [CrossRef]
- Chen, S.; Li, F.-Y.; Li, J.-F.; Sun, X.; Cui, J.-F.; Zhang, C.-W.; Wang, L.-M.; Xie, Q.; Xu, J. Effects of single-modification/cross-modification of starch on the mechanical properties of new biodegradable composites. RSC Adv. 2018, 8, 12400–12408. [Google Scholar] [CrossRef]
- Zha, X.; Sadi, M.S.; Yang, Y.; Luo, T.; Huang, N. Introduction of poly(acrylic acid) branch onto acetate starch for polyester warp sizing. J. Text. Inst. 2021, 112, 273–285. [Google Scholar] [CrossRef]
- Peidayesh, H.; Heydari, A.; Mosnáčková, K.; Chodák, I. In situ dual crosslinking strategy to improve the physico-chemical properties of thermoplastic starch. Carbohydr. Polym. 2021, 269, 118250. [Google Scholar] [CrossRef] [PubMed]
- Tessema, A.; Admassu, H.; Dereje, B. Development of edible films based on anchote (Coccinia abyssinica) starch: Process optimization using response surface methodology (RSM). J. Food Meas. Charact. 2023, 17, 430–446. [Google Scholar] [CrossRef]
- Pérez-Chávez, R.; Sánchez-Aguilar, J.; Calderas, F.; Maddalena, L.; Carosio, F.; Sanchez-Olivares, G. A statistical approach to the development of flame retardant and mechanically strong natural fibers biocomposites. Polym. Degrad. Stab. 2022, 201, 109991. [Google Scholar] [CrossRef]
- Luo, Y.; Xiao, S.; Li, S. Effect of initial water content on foaming quality and mechanical properties of plant fiber porous cushioning materials. BioResources 2017, 12, 4259–4269. [Google Scholar] [CrossRef]
- Singh, S.; Chakraborty, J.P.; Mondal, M.K. Pyrolysis of torrefied biomass: Optimization of process parameters using response surface methodology, characterization, and comparison of properties of pyrolysis oil from raw biomass. J. Clean. Prod. 2020, 272, 122517. [Google Scholar] [CrossRef]
- Xu, Y.-Z.; Soloway, R.D.; Lin, X.-F.; Zhi, X.; Weng, S.-F.; Wu, Q.-G.; Shi, J.-S.; Sun, W.-X.; Zhang, T.-X.; Wu, J.-G.; et al. Fourier transform infrared (FT-IR) mid-IR spectroscopy separates normal and malignant tissue from the colon and stomach. Gastroenterology 2000, 118, A1417. [Google Scholar] [CrossRef]
- Ishkhanyan, A.M.; Krainov, V.P. Van der Waals Attraction of Hydrogen Atoms. J. Exp. Theor. Phys. 2021, 132, 892–896. [Google Scholar] [CrossRef]
- Khatun, A.; Waters, D.L.E.; Liu, L. A Review of Rice Starch Digestibility: Effect of Composition and Heat-Moisture Processing. Starch-Stärke 2019, 71, 1900090. [Google Scholar] [CrossRef]
- Shahbazi, M.; Majzoobi, M.; Farahnaky, A. Impact of shear force on functional properties of native starch and resulting gel and film. J. Food Eng. 2018, 223, 10–21. [Google Scholar] [CrossRef]
- Onyishi, H.O.; Oluah, C.K. Effect of stretch ratio on the induced crystallinity and mechanical properties of biaxially stretched PET. Phase Transit. 2020, 93, 924–934. [Google Scholar] [CrossRef]
- He, X.; Wu, S.; Fu, D.; Ni, J. Preparation of sodium carboxymethyl cellulose from paper sludge. J. Chem. Technol. Biotechnol. 2009, 84, 427–434. [Google Scholar] [CrossRef]
- Agrawal, R.; Saxena, N.S.; Sharma, K.B.; Thomas, S.; Sreekala, M.S. Activation energy and crystallization kinetics of untreated and treated oil palm fibre reinforced phenol formaldehyde composites. Mater. Sci. Eng. A 2000, 277, 77–82. [Google Scholar] [CrossRef]
- Atapour, M.; Kariminia, H.-R.; Moslehabadi, P.M. Optimization of biodiesel production by alkali-catalyzed transesterification of used frying oil. Process Saf. Environ. Prot. 2014, 92, 179–185. [Google Scholar] [CrossRef]
- Azizi Namaghi, H.; Mousavi, S.M. Factorial experimental design for treatment of an industrial wastewater using micellar-enhanced ultrafiltration. Desalination Water Treat. 2016, 57, 5416–5424. [Google Scholar] [CrossRef]
- Haji, A.; Ghaheh, F.S.; Indrie, L. Pomegranate fallen leaves as a source of natural dye for mordant-free dyeing of wool. Color. Technol. 2023, 139, 165–170. [Google Scholar] [CrossRef]
- Lesch, S.M. Sensor-directed response surface sampling designs for characterizing spatial variation in soil properties. Comput. Electron. Agric. 2005, 46, 153–179. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCS:Glycerol | NCS:D−Fructose | NCS:Glycerol:D−Fructose |
|---|---|---|
| 10:1 | 10:1 | 10:1:1 |
| 10:2 | 10:2 | 10:2:1 |
| 10:3 | 10:3 | |
| 10:4 | 10:4 |
| Level | Forming Temperature (°C) | Forming Time (min) | AC Content (g) |
|---|---|---|---|
| −1 | 195 °C | 4 | 0.5 |
| 0 | 205 °C | 5 | 1 |
| 1 | 215 °C | 6 | 1.5 |
| Experiment Number | Forming Temperature (°C) | Forming Time (min) | AC Content (g) | Tensile Strength (MPa) | Rebound Rate (%) |
|---|---|---|---|---|---|
| 1 | 215 | 6.00 | 1.00 | 1.90 ± 0.14 | 64.4 ± 1.3 |
| 2 | 195 | 6.00 | 1.00 | 4.02 ± 0.18 | 92.5 ± 2.1 |
| 3 | 195 | 5.00 | 0.50 | 3.76 ± 0.05 | 88.5 ± 0.9 |
| 4 | 195 | 4.00 | 1.00 | 3.33 ± 0.13 | 87.2 ± 1.1 |
| 5 | 205 | 6.00 | 0.50 | 3.4 ± 0.09 | 92.4 ± 1.2 |
| 6 | 215 | 4.00 | 1.00 | 3.11 ± 0.06 | 89.7 ± 0.4 |
| 7 | 205 | 5.00 | 1.00 | 3.76 ± 0.05 | 94.0 ± 0.6 |
| 8 | 195 | 5.00 | 1.50 | 2.88 ± 0.08 | 91.6 ± 0.6 |
| 9 | 205 | 6.00 | 1.50 | 3.06 ± 0.3 | 73.3 ± 2.5 |
| 10 | 205 | 5.00 | 1.00 | 4.18 ± 0.08 | 95.7 ± 0.5 |
| 11 | 205 | 5.00 | 1.00 | 4.10 ± 0.07 | 94.5 ± 0.4 |
| 12 | 205 | 5.00 | 1.00 | 3.84 ± 0.09 | 93.7 ± 0.7 |
| 13 | 205 | 4.00 | 1.50 | 1.93 ± 0.03 | 84.8 ± 0.2 |
| 14 | 205 | 5.00 | 1.00 | 4.23 ± 0.4 | 89.4 ± 2.7 |
| 15 | 215 | 5.00 | 1.50 | 2.49 ± 0.07 | 64.2 ± 0.4 |
| 16 | 215 | 5.00 | 0.50 | 2.79 ± 0.20 | 73.3 ± 1.7 |
| 17 | 205 | 4.00 | 0.50 | 3.31 ± 0.08 | 92.1 ± 0.6 |
| Coefficient Source | Sum of Squares | Degree of Freedom | Variance | p−Value |
|---|---|---|---|---|
| model | 7.84 | 9 | 0.87 | 0.0049 |
| A− Forming temperature | 1.71 | 1 | 1.71 | 0.0045 |
| B− Forming time | 0.061 | 1 | 0.061 | 0.4629 |
| C−AC content | 1.05 | 1 | 1.05 | 0.0147 |
| AB | 0.90 | 1 | 0.90 | 0.0205 |
| AC | 0.084 | 1 | 0.084 | 0.3931 |
| BC | 0.27 | 1 | 0.27 | 0.1468 |
| A2 | 0.81 | 1 | 0.81 | 0.0257 |
| B2 | 1.03 | 1 | 1.03 | 0.0155 |
| C2 | 1.53 | 1 | 1.53 | 0.0060 |
| Residual error | 0.71 | 7 | 0.10 | 0.1050 |
| LOF | 0.53 | 3 | 0.18 | |
| Error term | 0.18 | 4 | 0.044 | |
| Total variation | 8.55 | 16 |
| Coefficient Source | Sum of Squares | Degree of Freedom | Variance | p−Value |
|---|---|---|---|---|
| model | 0.16 | 9 | 0.018 | 0.0027 |
| A− Forming temperature | 0.058 | 1 | 0.058 | 0.0007 |
| B− Forming time | 0.012 | 1 | 0.012 | 0.0327 |
| C−AC content | 0.013 | 1 | 0.013 | 0.0282 |
| AB | 0.024 | 1 | 0.024 | 0.0078 |
| AC | 0.0037 | 1 | 0.0037 | 0.1854 |
| BC | 0.0035 | 1 | 0.0035 | 0.1984 |
| A2 | 0.028 | 1 | 0.028 | 0.0051 |
| B2 | 0.0015 | 1 | 0.0015 | 0.3839 |
| C2 | 0.015 | 1 | 0.015 | 0.0220 |
| Residual error | 0.012 | 7 | 0.0017 | |
| LOF | 0.0098 | 3 | 0.0033 | 0.0632 |
| Error term | 0.0022 | 4 | 0.0057 | |
| Total variation | 0.17 | 16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Tang, R.; Guo, A.; Tao, X.; Hu, Y.; Sheng, X.; Qu, P.; Wang, S.; Li, J.; Li, F. Enhancing Starch−Based Packaging Materials: Optimization of Plasticizers and Process Parameters. Materials 2023, 16, 5953. https://doi.org/10.3390/ma16175953
Wu Y, Tang R, Guo A, Tao X, Hu Y, Sheng X, Qu P, Wang S, Li J, Li F. Enhancing Starch−Based Packaging Materials: Optimization of Plasticizers and Process Parameters. Materials. 2023; 16(17):5953. https://doi.org/10.3390/ma16175953
Chicago/Turabian StyleWu, Yue, Rongji Tang, Anfu Guo, Xiaodong Tao, Yingbin Hu, Xianliang Sheng, Peng Qu, Shaoqing Wang, Jianfeng Li, and Fangyi Li. 2023. "Enhancing Starch−Based Packaging Materials: Optimization of Plasticizers and Process Parameters" Materials 16, no. 17: 5953. https://doi.org/10.3390/ma16175953
APA StyleWu, Y., Tang, R., Guo, A., Tao, X., Hu, Y., Sheng, X., Qu, P., Wang, S., Li, J., & Li, F. (2023). Enhancing Starch−Based Packaging Materials: Optimization of Plasticizers and Process Parameters. Materials, 16(17), 5953. https://doi.org/10.3390/ma16175953