
Computational Exploration of Phenolic Compounds in Corrosion Inhibition: A Case Study of Hydroxytyrosol and Tyrosol


Abstract
:1. Introduction
2. Computational Details
2.1. Quantum Chemical Calculations
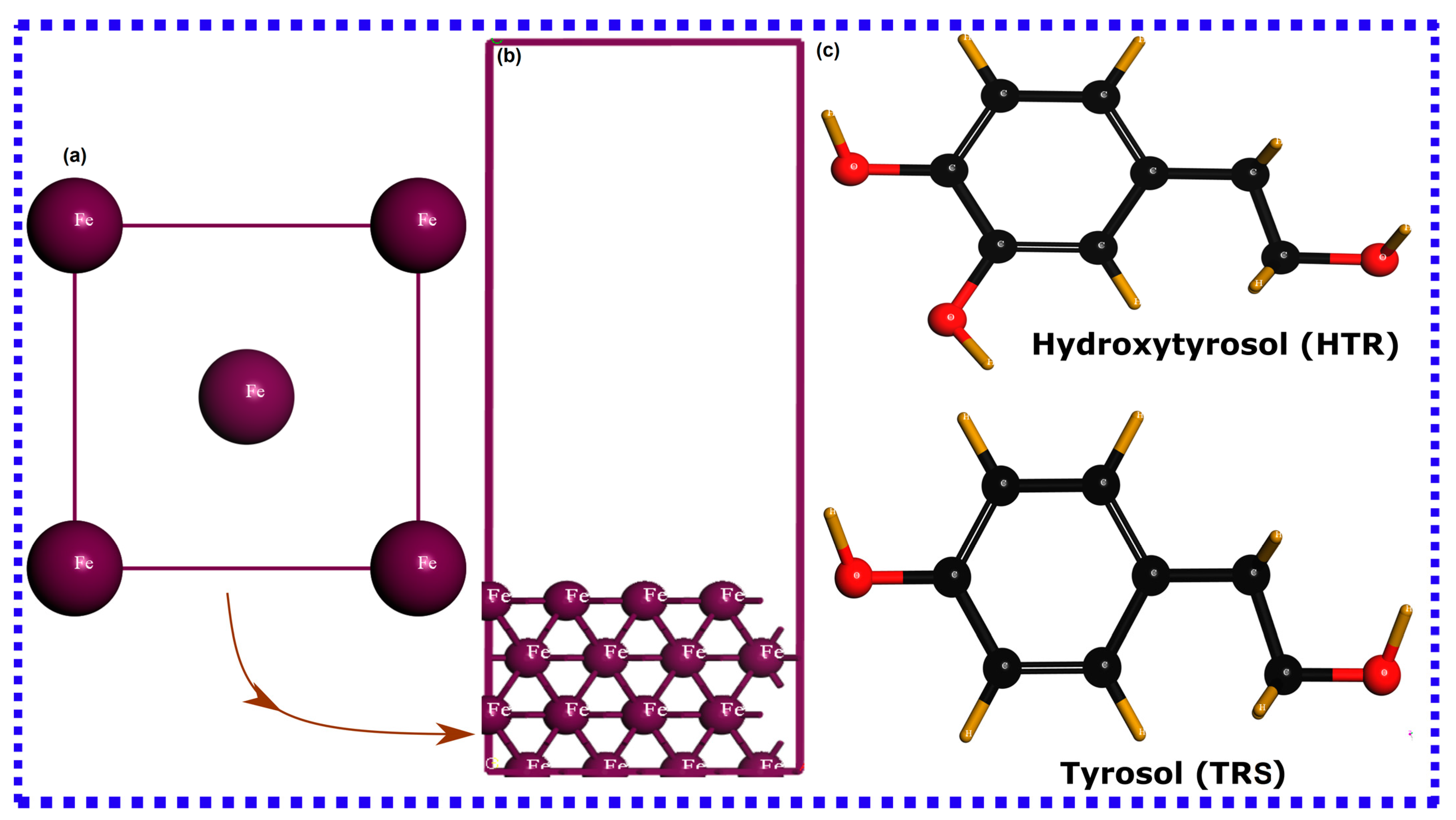
2.2. Molecular Dynamics Simulation
2.3. SCC-DFTB Computational Details
3. Results and Discussion
3.1. Quantum Chemical Calculations
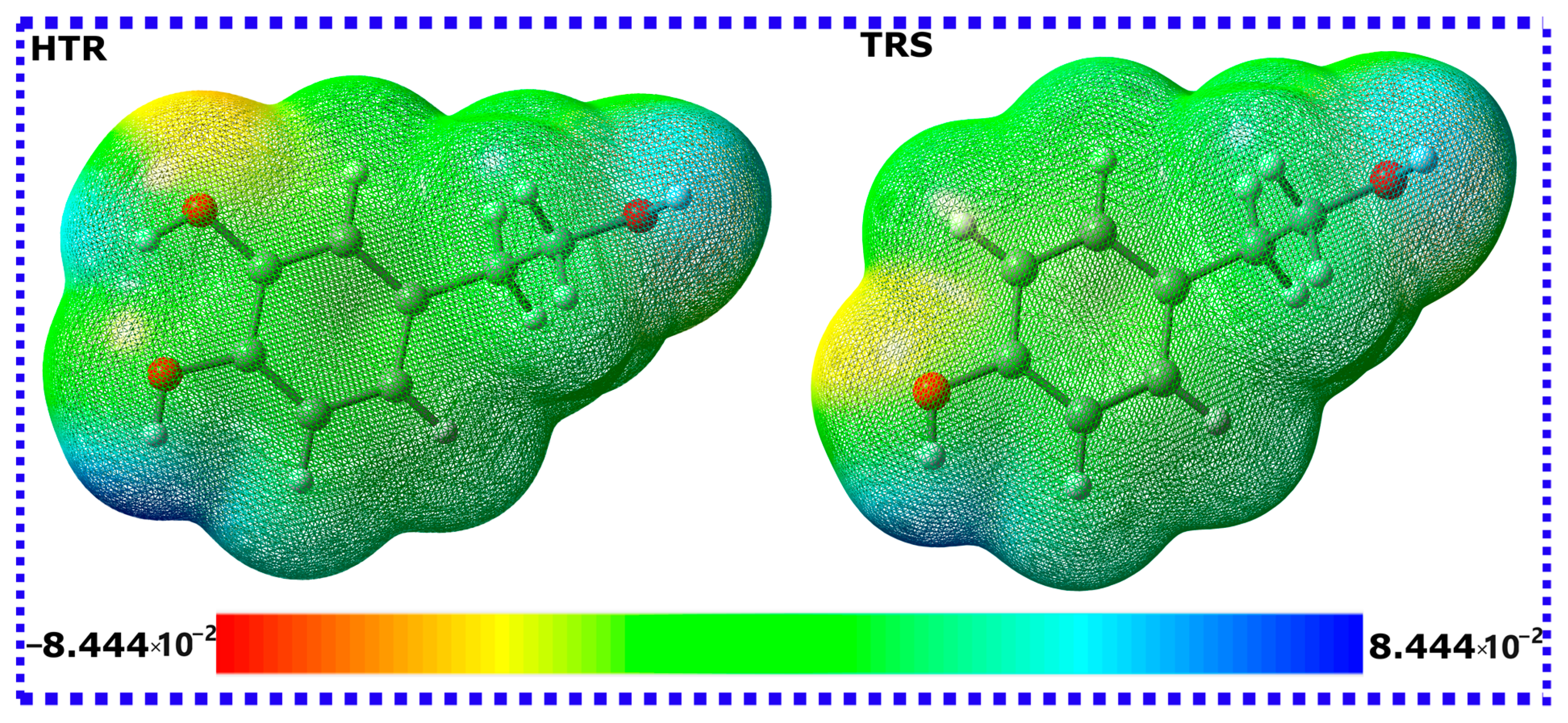
3.1.1. Global Reactivity Descriptors
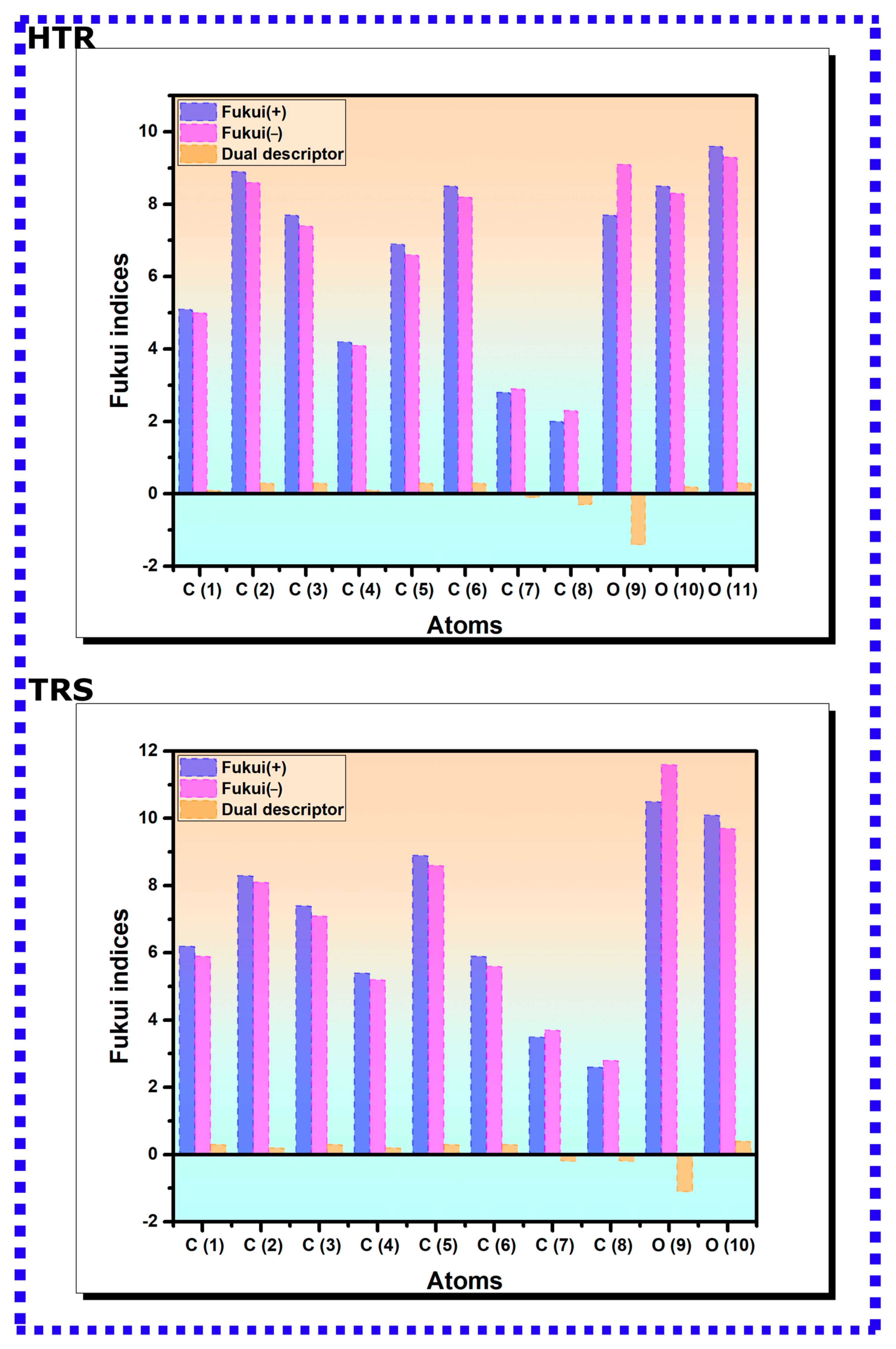
3.1.2. Fukui Function Indices
3.2. Semi-Empirical SCC-DFTB Calculations
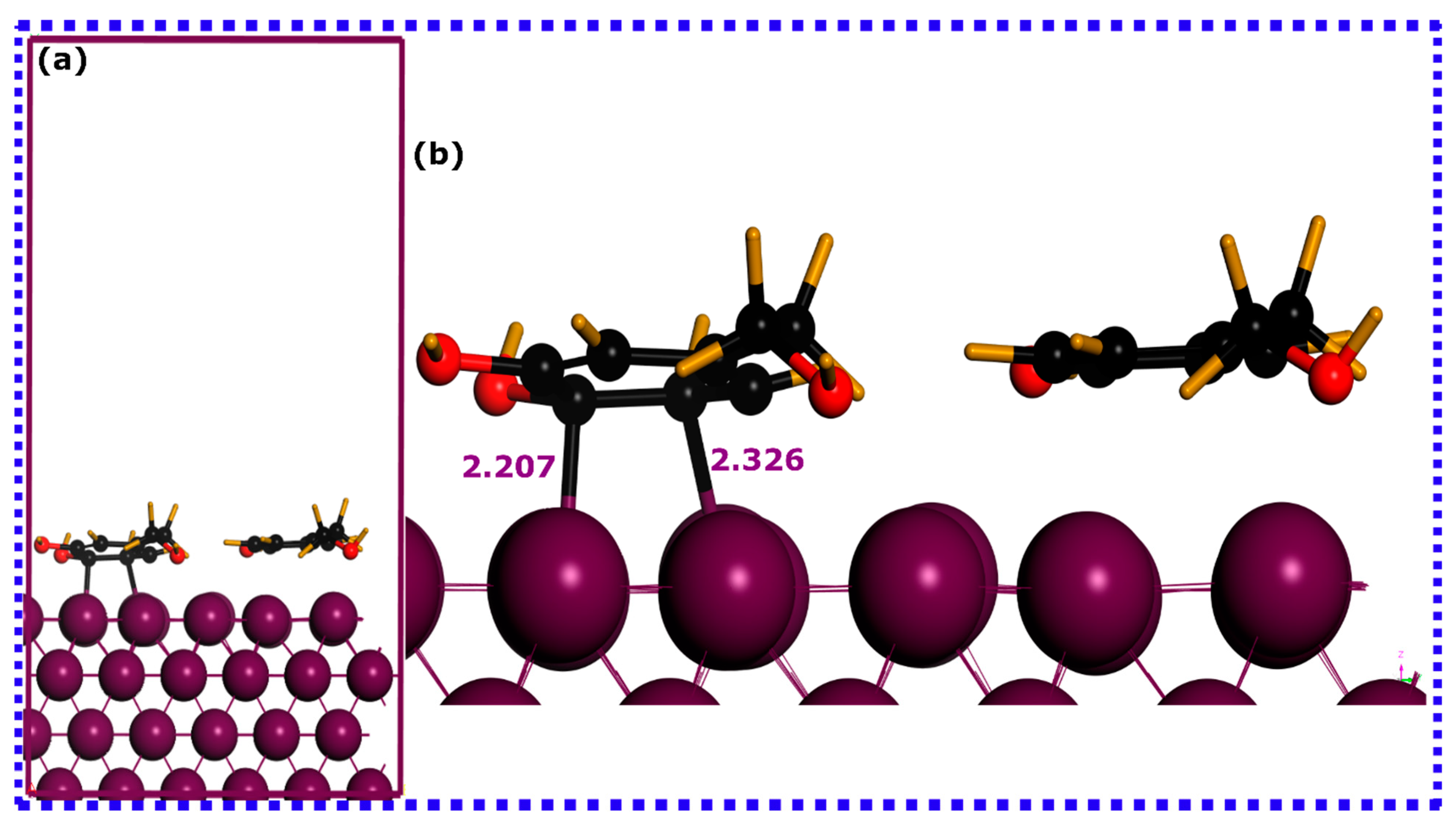
3.2.1. Optimized Geometries and Interaction Energies
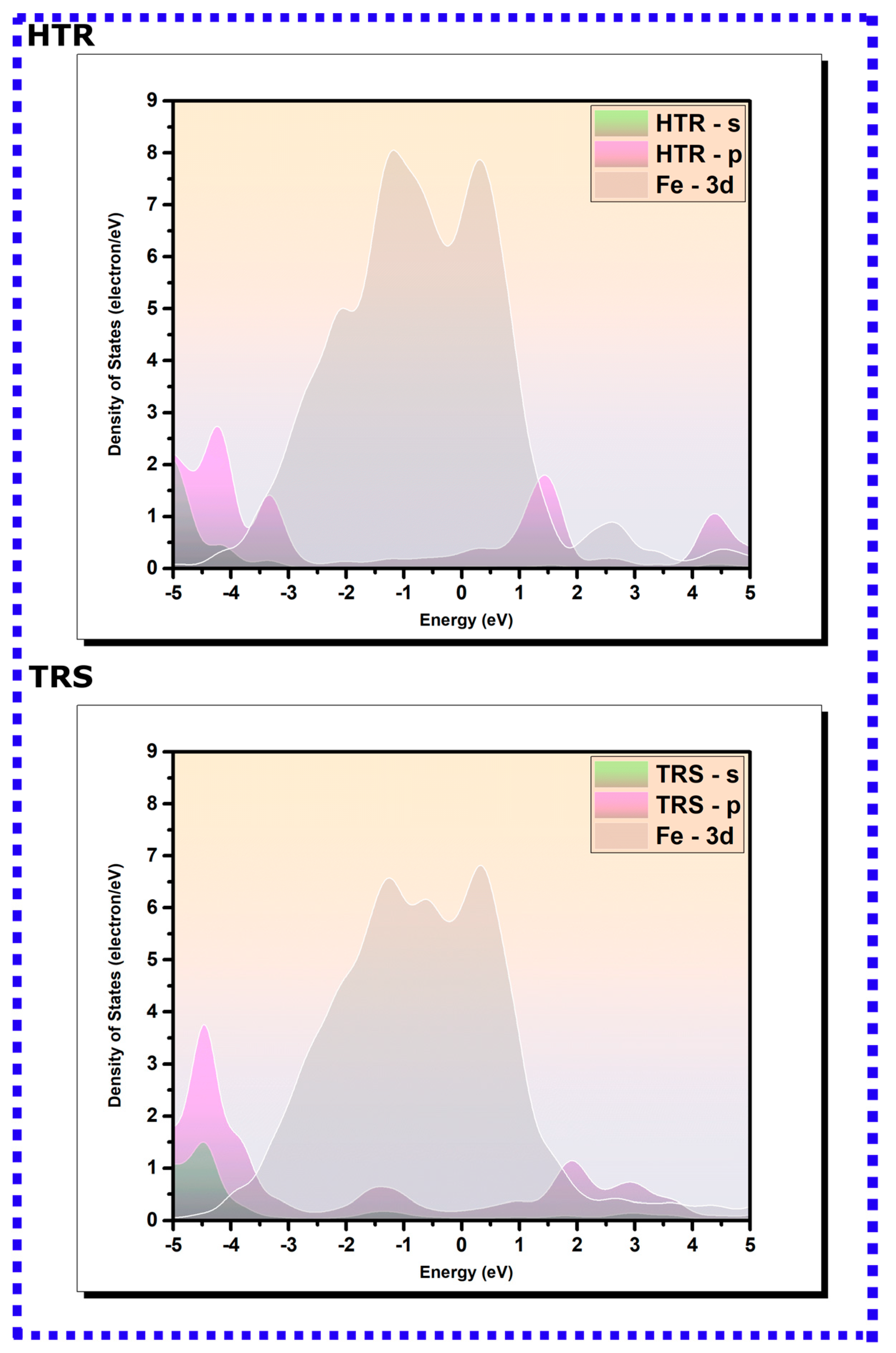
3.2.2. PDOS Analysis
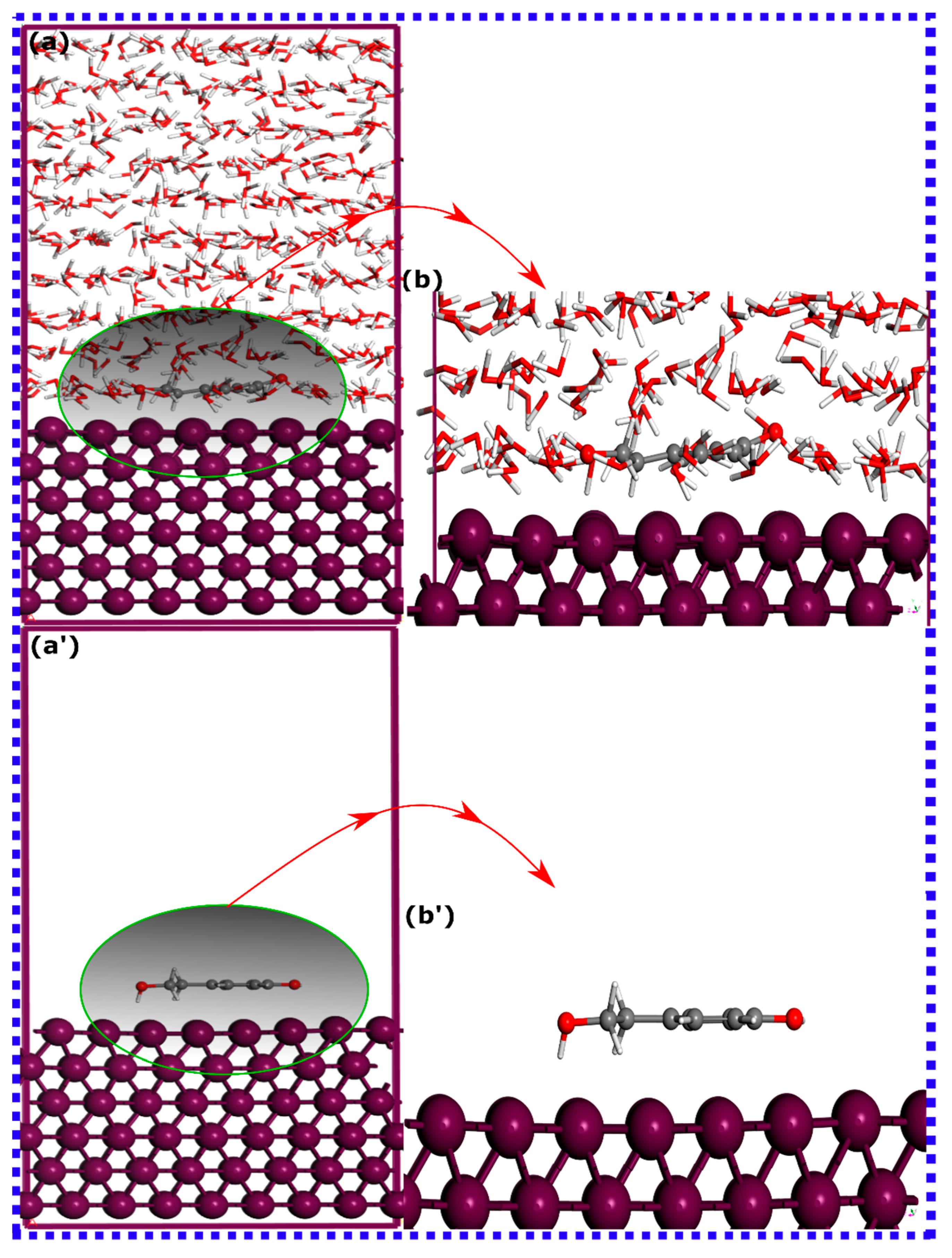
3.3. MD Simulations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El-Etre, A.Y. Inhibition of Acid Corrosion of Carbon Steel Using Aqueous Extract of Olive Leaves. J. Colloid Interface Sci. 2007, 314, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Ben Harb, M.; Abubshait, S.; Etteyeb, N.; Kamoun, M.; Dhouib, A. Olive Leaf Extract as a Green Corrosion Inhibitor of Reinforced Concrete Contaminated with Seawater. Arab. J. Chem. 2020, 13, 4846–4856. [Google Scholar] [CrossRef]
- Bouknana, D.; Hammouti, B.; Serghini Caid, H.; Jodeh, S.; Bouyanzer, A.; Aouniti, A.; Warad, I. Aqueous Extracts of Olive Roots, Stems, and Leaves as Eco-Friendly Corrosion Inhibitor for Steel in 1 MHCl Medium. Int. J. Ind. Chem. 2015, 6, 233–245. [Google Scholar] [CrossRef]
- Marder, A.R.; Goodwin, F.E. Chapter 11—Corrosion Behavior. In The Metallurgy of Zinc Coated Steels; Marder, A.R., Goodwin, F.E., Eds.; Woodhead Publishing Series in Metals and Surface Engineering; Elsevier: Amsterdam, The Netherlands, 2023; pp. 341–396. ISBN 978-0-323-99984-7. [Google Scholar]
- Thakur, A.; Assad, H.; Kaya, S.; Kumar, A. Chapter 30—Plant Extracts as Bio-Based Anticorrosive Materials. In Handbook of Biomolecules; Verma, C., Verma, D.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2023; pp. 591–618. ISBN 978-0-323-91684-4. [Google Scholar]
- Haldhar, R.; Kim, S.-C.; Dagdag, O.; Verma, D.K. Chapter 26—Amino Acids and Nucleic Acids as Green Corrosion Inhibitors. In Handbook of Biomolecules; Verma, C., Verma, D.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2023; pp. 523–533. ISBN 978-0-323-91684-4. [Google Scholar]
- Karković Marković, A.; Torić, J.; Barbarić, M.; Jakobušić Brala, C. Hydroxytyrosol, Tyrosol and Derivatives and Their Potential Effects on Human Health. Molecules 2019, 24, 2001. [Google Scholar] [CrossRef]
- Rodríguez-Morató, J.; Boronat, A.; Kotronoulas, A.; Pujadas, M.; Pastor, A.; Olesti, E.; Pérez-Mañá, C.; Khymenets, O.; Fitó, M.; Farré, M.; et al. Metabolic Disposition and Biological Significance of Simple Phenols of Dietary Origin: Hydroxytyrosol and Tyrosol. Drug Metab. Rev. 2016, 48, 218–236. [Google Scholar] [CrossRef]
- Lucci, P.; Bertoz, V.; Pacetti, D.; Moret, S.; Conte, L. Effect of the Refining Process on Total Hydroxytyrosol, Tyrosol, and Tocopherol Contents of Olive Oil. Foods 2020, 9, 292. [Google Scholar] [CrossRef] [PubMed]
- Mateos, R.; Goya, L.; Bravo, L. Metabolism of the Olive Oil Phenols Hydroxytyrosol, Tyrosol, and Hydroxytyrosyl Acetate by Human Hepatoma HepG2 Cells. J. Agric. Food Chem. 2005, 53, 9897–9905. [Google Scholar] [CrossRef]
- Bordiga, M.; Lorenzo, C.; Pardo, F.; Salinas, M.R.; Travaglia, F.; Arlorio, M.; Coïsson, J.D.; Garde-Cerdán, T. Factors Influencing the Formation of Histaminol, Hydroxytyrosol, Tyrosol, and Tryptophol in Wine: Temperature, Alcoholic Degree, and Amino Acids Concentration. Food Chem. 2016, 197, 1038–1045. [Google Scholar] [CrossRef]
- Anter, J.; Tasset, I.; Demyda-Peyrás, S.; Ranchal, I.; Moreno-Millán, M.; Romero-Jimenez, M.; Muntané, J.; Luque de Castro, M.D.; Muñoz-Serrano, A.; Alonso-Moraga, Á. Evaluation of Potential Antigenotoxic, Cytotoxic and Proapoptotic Effects of the Olive Oil by-Product “Alperujo”, Hydroxytyrosol, Tyrosol and Verbascoside. Mutat. Res./Genet. Toxicol. Environ. Mutagen. 2014, 772, 25–33. [Google Scholar] [CrossRef]
- Abdel-Gaber, A.M.; Abd-El-Nabey, B.A.; Khamis, E.; Abd-El-Khalek, D.E. A Natural Extract as Scale and Corrosion Inhibitor for Steel Surface in Brine Solution. Desalination 2011, 278, 337–342. [Google Scholar] [CrossRef]
- Essien, E.A.; Kavaz, D.; Ituen, E.B.; Umoren, S.A. Synthesis, Characterization and Anticorrosion Property of Olive Leaves Extract-Titanium Nanoparticles Composite. J. Adhes. Sci. Technol. 2018, 32, 1773–1794. [Google Scholar] [CrossRef]
- Rahal, C.; Masmoudi, M.; Abdelhedi, R.; Sabot, R.; Jeannin, M.; Bouaziz, M.; Refait, P. Olive Leaf Extract as Natural Corrosion Inhibitor for Pure Copper in 0.5M NaCl Solution: A Study by Voltammetry around OCP. J. Electroanal. Chem. 2016, 769, 53–61. [Google Scholar] [CrossRef]
- Wei, H.; Heidarshenas, B.; Zhou, L.; Hussain, G.; Li, Q.; Ostrikov, K. (Ken) Green Inhibitors for Steel Corrosion in Acidic Environment: State of Art. Mater. Today Sustain. 2020, 10, 100044. [Google Scholar] [CrossRef]
- Medupin, R.O.; Ukoba, K.O.; Yoro, K.O.; Jen, T.-C. Sustainable Approach for Corrosion Control in Mild Steel Using Plant-Based Inhibitors: A Review. Mater. Today Sustain. 2023, 22, 100373. [Google Scholar] [CrossRef]
- Al Jahdaly, B.A.; Maghraby, Y.R.; Ibrahim, A.H.; Shouier, K.R.; Alturki, A.M.; El-Shabasy, R.M. Role of Green Chemistry in Sustainable Corrosion Inhibition: A Review on Recent Developments. Mater. Today Sustain. 2022, 20, 100242. [Google Scholar] [CrossRef]
- Ahmed, M.; Khan, K.-R.; Ahmad, S.; Aati, H.Y.; Sherif, A.E.; Ashkan, M.F.; Alrahimi, J.; Abdullah Motwali, E.; Imran Tousif, M.; Abbas Khan, M.; et al. Phytochemical, Antioxidant, Enzyme Inhibitory, Thrombolytic, Antibacterial, Antiviral and in Silico Studies of Acacia Jacquemontii Leaves. Arab. J. Chem. 2022, 15, 104345. [Google Scholar] [CrossRef]
- Fernandes, C.M.; Ferreira Fagundes, T.d.S.; Escarpini dos Santos, N.; Shewry, D.M.; Rocha, T.; Garrett, R.; Borges, R.M.; Muricy, G.; Valverde, A.L.; Ponzio, E.A. Ircinia Strobilina Crude Extract as Corrosion Inhibitor for Mild Steel in Acid Medium. Electrochim. Acta 2019, 312, 137–148. [Google Scholar] [CrossRef]
- Kathiravan, S.; Jyothi, S.; Ayyannan, G.; Ravichandran, J.; Raja, G. Inhibitory Action of Aqueous Ruellia Tuberosa L Leaves Extract on the Corrosion of Copper in HCl Solution. J. Indian Chem. Soc. 2021, 98, 100207. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, L.; Zhang, Q.; Wu, X.; Zheng, H.; Gao, P.; Zeng, G.; Yan, Z.; Sun, Y.; Li, Z.; et al. Insight into the Anti–Corrosion Performance of Artemisia Argyi Leaves Extract as Eco–Friendly Corrosion Inhibitor for Carbon Steel in HCl Medium. Sustain. Chem. Pharm. 2022, 27, 100710. [Google Scholar] [CrossRef]
- Wang, W.; Song, Z.; Guo, M.; Jiang, L.; Xiao, B.; Jiang, Q.; Chu, H.; Liu, Y.; Zhang, Y.; Xu, N. Employing Ginger Extract as an Eco-Friendly Corrosion Inhibitor in Cementitious Materials. Constr. Build. Mater. 2019, 228, 116713. [Google Scholar] [CrossRef]
- Shahmoradi, A.R.; Ranjbarghanei, M.; Javidparvar, A.A.; Guo, L.; Berdimurodov, E.; Ramezanzadeh, B. Theoretical and Surface/Electrochemical Investigations of Walnut Fruit Green Husk Extract as Effective Inhibitor for Mild-Steel Corrosion in 1M HCl Electrolyte. J. Mol. Liq. 2021, 338, 116550. [Google Scholar] [CrossRef]
- Sanaei, Z.; Bahlakeh, G.; Ramezanzadeh, B.; Ramezanzadeh, M. Application of Green Molecules from Chicory Aqueous Extract for Steel Corrosion Mitigation against Chloride Ions Attack; the Experimental Examinations and Electronic/Atomic Level Computational Studies. J. Mol. Liq. 2019, 290, 111176. [Google Scholar] [CrossRef]
- Ramezanzadeh, M.; Bahlakeh, G.; Ramezanzadeh, B. Study of the Synergistic Effect of Mangifera Indica Leaves Extract and Zinc Ions on the Mild Steel Corrosion Inhibition in Simulated Seawater: Computational and Electrochemical Studies. J. Mol. Liq. 2019, 292, 111387. [Google Scholar] [CrossRef]
- Dehghani, A.; Bahlakeh, G.; Ramezanzadeh, B. Green Eucalyptus Leaf Extract: A Potent Source of Bio-Active Corrosion Inhibitors for Mild Steel. Bioelectrochemistry 2019, 130, 107339. [Google Scholar] [CrossRef]
- Lgaz, H.; Chaouiki, A.; Lamouri, R.; Salghi, R.; Lee, H.-S. Computational Methods of Corrosion Inhibition Assessment. In Sustainable Corrosion Inhibitors I: Fundamentals, Methodologies, and Industrial Applications; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2021; Volume 1403, pp. 87–109. ISBN 978-0-8412-9790-6. [Google Scholar]
- Obot, I.B.; Haruna, K.; Saleh, T.A. Atomistic Simulation: A Unique and Powerful Computational Tool for Corrosion Inhibition Research. Arab. J. Sci. Eng. 2018, 44, 1–32. [Google Scholar] [CrossRef]
- Ebenso, E.E.; Verma, C.; Olasunkanmi, L.O.; Akpan, E.D.; Kumar, V.D.; Lgaz, H.; Guo, L.; Kaya, S.; Quraishi, M.A. Molecular Modelling of Compounds Used for Corrosion Inhibition Studies: A Review. Phys. Chem. Chem. Phys. 2021, 23, 19987–20027. [Google Scholar] [CrossRef]
- Wazzan, N. Phytochemical Components of Allium Jesdianum Flower as Effective Corrosion-Resistant Materials for Fe(110), Al(111), and Cu(111): DFT Study. Arab. J. Chem. 2023, 16, 104625. [Google Scholar] [CrossRef]
- Kumar, D.; Jain, V.; Rai, B. Unravelling the Mechanisms of Corrosion Inhibition of Iron by Henna Extract: A Density Functional Theory Study. Corros. Sci. 2018, 142, 102–109. [Google Scholar] [CrossRef]
- Alaoui, K.; Ouakki, M.; Abousalem, A.S.; Serrar, H.; Galai, M.; Derbali, S.; Nouneh, K.; Boukhris, S.; Touhami, M.E.; El Kacimi, Y. Molecular Dynamics, Monte-Carlo Simulations and Atomic Force Microscopy to Study the Interfacial Adsorption Behaviour of Some Triazepine Carboxylate Compounds as Corrosion Inhibitors in Acid Medium. J. Bio- Tribo-Corros. 2018, 5, 1. [Google Scholar] [CrossRef]
- Oukhrib, R.; Abdellaoui, Y.; Berisha, A.; Abou Oualid, H.; Halili, J.; Jusufi, K.; Ait El Had, M.; Bourzi, H.; El Issami, S.; Asmary, F.A. DFT, Monte Carlo and Molecular Dynamics Simulations for the Prediction of Corrosion Inhibition Efficiency of Novel Pyrazolylnucleosides on Cu(111) Surface in Acidic Media. Sci. Rep. 2021, 11, 3771. [Google Scholar] [CrossRef]
- Verma, C.; Lgaz, H.; Verma, D.K.; Ebenso, E.E.; Bahadur, I.; Quraishi, M.A. Molecular Dynamics and Monte Carlo Simulations as Powerful Tools for Study of Interfacial Adsorption Behavior of Corrosion Inhibitors in Aqueous Phase: A Review. J. Mol. Liq. 2018, 260, 99–120. [Google Scholar] [CrossRef]
- Kokalj, A.; Costa, D. Molecular Modeling of Corrosion Inhibitors. In Encyclopedia of Interfacial Chemistry; Wandelt, K., Ed.; Elsevier: Oxford, UK, 2018; pp. 332–345. ISBN 978-0-12-809894-3. [Google Scholar]
- Liu, H.; Seifert, G.; Di Valentin, C. An Efficient Way to Model Complex Magnetite: Assessment of SCC-DFTB against DFT. J. Chem. Phys. 2019, 150, 094703. [Google Scholar] [CrossRef] [PubMed]
- Seabra, G.d.M.; Walker, R.C.; Elstner, M.; Case, D.A.; Roitberg, A.E. Implementation of the SCC-DFTB Method for Hybrid QM/MM Simulations within the Amber Molecular Dynamics Package. J. Phys. Chem. A 2007, 111, 5655–5664. [Google Scholar] [CrossRef] [PubMed]
- El-Haitout, B.; Selatnia, I.; Lgaz, H.; Al-Hadeethi, M.R.; Lee, H.-S.; Chaouiki, A.; Ko, Y.G.; Ali, I.H.; Salghi, R. Exploring the Feasibility of New Eco-Friendly Heterocyclic Compounds for Establishing Efficient Corrosion Protection for N80 Steel in a Simulated Oil Well Acidizing Environment: From Molecular-Level Prediction to Experimental Validation. Colloids Surf. A Physicochem. Eng. Asp. 2023, 656, 130372. [Google Scholar] [CrossRef]
- Lgaz, H.; Lee, H. Facile Preparation of New Hydrazone Compounds and Their Application for Long-Term Corrosion Inhibition of N80 Steel in 15% HCl: An Experimental Study Combined with DFTB Calculations. J. Mol. Liq. 2022, 347, 117952. [Google Scholar] [CrossRef]
- Guo, L.; Qi, C.; Zheng, X.; Zhang, R.; Shen, X.; Kaya, S. Toward Understanding the Adsorption Mechanism of Large Size Organic Corrosion Inhibitors on an Fe(110) Surface Using the DFTB Method. RSC Adv. 2017, 7, 29042–29050. [Google Scholar] [CrossRef]
- Obot, I.B.; Macdonald, D.D.; Gasem, Z.M. Density Functional Theory (DFT) as a Powerful Tool for Designing New Organic Corrosion Inhibitors. Part 1: An Overview. Corros. Sci. 2015, 99, 1–30. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Delley, B. An All-electron Numerical Method for Solving the Local Density Functional for Polyatomic Molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. From Molecules to Solids with the DMol 3 Approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Ansari, A.; Ou-Ani, O.; Oucheikh, L.; Youssefi, Y.; Chebabe, D.; Oubair, A.; Znini, M. Experimental, Theoretical Modeling and Optimization of Inhibitive Action of Ocimum Basilicum Essential Oil as Green Corrosion Inhibitor for C38 Steel in 0.5 MH2SO4 Medium. Chem. Afr. 2022, 5, 37–55. [Google Scholar] [CrossRef]
- Saha, S.K.; Dutta, A.; Ghosh, P.; Sukul, D.; Banerjee, P. Novel Schiff-Base Molecules as Efficient Corrosion Inhibitors for Mild Steel Surface in 1 M HCl Medium: Experimental and Theoretical Approach. Phys. Chem. Chem. Phys. 2016, 18, 17898–17911. [Google Scholar] [CrossRef] [PubMed]
- Frisch, A. Gaussian 09W Reference; Gaussian, Inc.: Wallingford, UK, 2009; 25p. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView 6.0. 16; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Akkermans, R.L.; Spenley, N.A.; Robertson, S.H. COMPASS III: Automated Fitting Workflows and Extension to Ionic Liquids. Mol. Simul. 2021, 47, 540–551. [Google Scholar] [CrossRef]
- Paterlini, M.G.; Ferguson, D.M. Constant Temperature Simulations Using the Langevin Equation with Velocity Verlet Integration. Chem. Phys. 1998, 236, 243–252. [Google Scholar] [CrossRef]
- Evans, D.J.; Holian, B.L. The Nose–Hoover Thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Biovia, D.S. Materials Studio, Version 6.0; Accelrys Inc.: San Diego, CA, USA, 2012. [Google Scholar]
- Zheng, G.; Witek, H.A.; Bobadova-Parvanova, P.; Irle, S.; Musaev, D.G.; Prabhakar, R.; Morokuma, K.; Lundberg, M.; Elstner, M.; Köhler, C. Parameter Calibration of Transition-Metal Elements for the Spin-Polarized Self-Consistent-Charge Density-Functional Tight-Binding (DFTB) Method: Sc, Ti, Fe, Co, and Ni. J. Chem. Theory Comput. 2007, 3, 1349–1367. [Google Scholar] [CrossRef]
- Bai, J.; Liu, X.; Guo, W.; Lei, T.; Teng, B.; Xiang, H.; Wen, X. An Efficient Way to Model Complex Iron Carbides: A Benchmark Study of DFTB2 against DFT. J. Phys. Chem. A 2023, 127, 2071–2080. [Google Scholar] [CrossRef]
- Liu, C.; Batista, E.R.; Aguirre, N.F.; Yang, P.; Cawkwell, M.J.; Jakubikova, E. SCC-DFTB Parameters for Fe–C Interactions. J. Phys. Chem. A 2020, 124, 9674–9682. [Google Scholar] [CrossRef]
- Hourahine, B.; Aradi, B.; Blum, V.; Bonafé, F.; Buccheri, A.; Camacho, C.; Cevallos, C.; Deshaye, M.; Dumitrică, T.; Dominguez, A. DFTB+, a Software Package for Efficient Approximate Density Functional Theory Based Atomistic Simulations. J. Chem. Phys. 2020, 152, 124101. [Google Scholar] [CrossRef]
- Isin, D.O.; Karakus, N. Quantum Chemical Study on the Inhibition Efficiencies of Some Sym-Triazines as Inhibitors for Mild Steel in Acidic Medium. J. Taiwan Inst. Chem. Eng. 2015, 50, 306–313. [Google Scholar] [CrossRef]
- Saha, S.K.; Murmu, M.; Murmu, N.C.; Obot, I.B.; Banerjee, P. Molecular Level Insights for the Corrosion Inhibition Effectiveness of Three Amine Derivatives on the Carbon Steel Surface in the Adverse Medium: A Combined Density Functional Theory and Molecular Dynamics Simulation Study. Surf. Interfaces 2018, 10, 65–73. [Google Scholar] [CrossRef]
- Islam, N.; Kaya, S. Conceptual Density Functional Theory and Its Application in the Chemical Domain; CRC Press: Boca Raton, FL, USA, 2018; ISBN 978-1-351-36024-1. [Google Scholar]
- Kokalj, A. Comments on the “Reply to Comments on the Paper ‘On the Nature of Inhibition Performance of Imidazole on Iron Surface’” by JO Mendes and AB Rocha. Corros. Sci. 2013, 70, 294–297. [Google Scholar] [CrossRef]
- Kokalj, A. On the HSAB Based Estimate of Charge Transfer between Adsorbates and Metal Surfaces. Chem. Phys. 2012, 393, 1–12. [Google Scholar] [CrossRef]
- Kokalj, A.; Lozinšek, M.; Kapun, B.; Taheri, P.; Neupane, S.; Losada-Pérez, P.; Xie, C.; Stavber, S.; Crespo, D.; Renner, F.U. Simplistic Correlations between Molecular Electronic Properties and Inhibition Efficiencies: Do They Really Exist? Corros. Sci. 2021, 179, 108856. [Google Scholar] [CrossRef]
- Kokalj, A. Is the Analysis of Molecular Electronic Structure of Corrosion Inhibitors Sufficient to Predict the Trend of Their Inhibition Performance. Electrochim. Acta 2010, 56, 745–755. [Google Scholar] [CrossRef]
- Thanikaivelan, P.; Padmanabhan, J.; Subramanian, V.; Ramasami, T. Chemical Reactivity and Selectivity Using Fukui Functions: Basis Set and Population Scheme Dependence in the Framework of B3LYP Theory. Theor. Chem. Acc. 2002, 107, 326–335. [Google Scholar] [CrossRef]
- Missioui, M.; Lgaz, H.; Guerrab, W.; Lee, H.; Warad, I.; Mague, J.T.; Ali, I.H.; Essassi, E.M.; Ramli, Y. Synthesis of Novel Hybrid Quinoxaline Containing Triazole and Acetamide Moieties by Azide-Alkyne Click Chemistry: Experimental and Theoretical Characterization. J. Mol. Struct. 2022, 1253, 132132. [Google Scholar] [CrossRef]
- Dutta, A.; Saha, S.K.; Banerjee, P.; Sukul, D. Correlating Electronic Structure with Corrosion Inhibition Potentiality of Some Bis-Benzimidazole Derivatives for Mild Steel in Hydrochloric Acid: Combined Experimental and Theoretical Studies. Corros. Sci. 2015, 98, 541–550. [Google Scholar] [CrossRef]
- Makedonas, C.; Mitsopoulou, C.A. An Investigation of the Reactivity of [(Diimine)(Dithiolato)M] Complexes Using the Fukui Functions Concept. Eur. J. Inorg. Chem. 2006, 2006, 590–598. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. New Dual Descriptor for Chemical Reactivity. J. Phys. Chem. A 2005, 109, 205–212. [Google Scholar] [CrossRef]
- Suresh, C.H.; Remya, G.S.; Anjalikrishna, P.K. Molecular Electrostatic Potential Analysis: A Powerful Tool to Interpret and Predict Chemical Reactivity. WIREs Comput. Mol. Sci. 2022, 12, e1601. [Google Scholar] [CrossRef]
- Bayoumy, A.M.; Ibrahim, M.; Omar, A. Mapping Molecular Electrostatic Potential (MESP) for Fulleropyrrolidine and Its Derivatives. Opt. Quant. Electron. 2020, 52, 346. [Google Scholar] [CrossRef]
- Oyeneyin, O.E.; Ojo, N.D.; Ipinloju, N.; James, A.C.; Agbaffa, E.B. Investigation of Corrosion Inhibition Potentials of Some Aminopyridine Schiff Bases Using Density Functional Theory and Monte Carlo Simulation. Chem. Afr. 2022, 5, 319–332. [Google Scholar] [CrossRef]
- Verma, C.; Olasunkanmi, L.O.; Ebenso, E.E.; Quraishi, M.A. Substituents Effect on Corrosion Inhibition Performance of Organic Compounds in Aggressive Ionic Solutions: A Review. J. Mol. Liq. 2018, 251, 100–118. [Google Scholar] [CrossRef]
- Kokalj, A.; Kovačević, N.; Peljhan, S.; Finšgar, M.; Lesar, A.; Milošev, I. Triazole, Benzotriazole, and Naphthotriazole as Copper Corrosion Inhibitors: I. Molecular Electronic and Adsorption Properties. ChemPhysChem 2011, 12, 3547–3555. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Jain, V.; Rai, B. Capturing the Synergistic Effects between Corrosion Inhibitor Molecules Using Density Functional Theory and ReaxFF Simulations—A Case for Benzyl Azide and Butyn-1-Ol on Cu Surface. Corros. Sci. 2022, 195, 109960. [Google Scholar] [CrossRef]
- Cordero, B.; Gómez, V.; Platero-Prats, A.E.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent Radii Revisited. Dalton Trans. 2008, 21, 2832–2838. [Google Scholar] [CrossRef]
- Kumar, D.; Jain, V.; Rai, B. Imidazole Derivatives as Corrosion Inhibitors for Copper: A DFT and Reactive Force Field Study. Corros. Sci. 2020, 171, 108724. [Google Scholar] [CrossRef]
- Deka, H.; Saikia, M.D.; Srivastava, H.K. Adsorption of Various Monoterpenoids on the Surface of Graphene and Nitrogen-Doped Graphene: A DFT Based Study. ChemistrySelect 2017, 2, 5248–5258. [Google Scholar] [CrossRef]
- Prates Ramalho, J.P.; Illas, F. Assessing the Importance of Van Der Waals Interactions on the Adsorption of Azobenzene on the Rutile TiO2(110) Surface. Chem. Phys. Lett. 2012, 545, 60–65. [Google Scholar] [CrossRef]
- Solmaz, R. Investigation of Adsorption and Corrosion Inhibition of Mild Steel in Hydrochloric Acid Solution by 5-(4-Dimethylaminobenzylidene)Rhodanine. Corros. Sci. 2014, 79, 169–176. [Google Scholar] [CrossRef]
- Solmaz, R. Investigation of the Inhibition Effect of 5-((E)-4-Phenylbuta-1,3-Dienylideneamino)-1,3,4-Thiadiazole-2-Thiol Schiff Base on Mild Steel Corrosion in Hydrochloric Acid. Corros. Sci. 2010, 52, 3321–3330. [Google Scholar] [CrossRef]
- Yüce, A.O.; Solmaz, R.; Kardaş, G. Investigation of Inhibition Effect of Rhodanine-N-Acetic Acid on Mild Steel Corrosion in HCl Solution. Mater. Chem. Phys. 2012, 131, 615–620. [Google Scholar] [CrossRef]
- Khnifira, M.; El Hamidi, S.; Sadiq, M.; Şimşek, S.; Kaya, S.; Barka, N.; Abdennouri, M. Adsorption Mechanisms Investigation of Methylene Blue on the (001) Zeolite 4A Surface in Aqueous Medium by Computational Approach and Molecular Dynamics. Appl. Surf. Sci. 2022, 572, 151381. [Google Scholar] [CrossRef]
- El-Haitout, B.; Hejjaj, C.; Lgaz, H.; Al-Hadeethi, M.R.; Ali, O.M.; Lee, H.-S.; Ali, I.H.; Salghi, R. Superior Long-Term Corrosion Inhibition of N80 Steel by New Eco-Friendly Hydrazone-Based Compounds in a Simulated Oil Well Acidizing Environment: Establishing the Mechanism at the Molecular Level. Langmuir 2022, 38, 15937–15949. [Google Scholar] [CrossRef] [PubMed]
- Tielens, F.; Santos, E. AuS and SH Bond Formation/Breaking during the Formation of Alkanethiol SAMs on Au(111): A Theoretical Study. J. Phys. Chem. C 2010, 114, 9444–9452. [Google Scholar] [CrossRef]
- Machado Fernandes, C.; Guedes, L.; Alvarez, L.X.; Barrios, A.M.; Lgaz, H.; Lee, H.-S.; Ponzio, E.A. Anticorrosive Properties of Green-Synthetized Benzylidene Derivatives for Mild Steel in Hydrochloric Acid: An Experimental Study Combined with DFTB and Molecular Dynamics Simulations. J. Mol. Liq. 2022, 363, 119790. [Google Scholar] [CrossRef]
- Chafiq, M.; Thari, F.Z.; Lee, H.; Chaouiki, A.; Salghi, R.; Ko, Y.G.; Karrouchi, K.; Bougrin, K.; Ali, I.H.; Lgaz, H. Experimental and First-Principles DFT Insights into the Corrosion Protection Mechanism of Carbon Steel in an HCl Medium by Two Thiazolidinedione Compounds. Mater. Today Commun. 2022, 32, 103841. [Google Scholar] [CrossRef]
- Thomas, A.G.; Jackman, M.J.; Wagstaffe, M.; Radtke, H.; Syres, K.; Adell, J.; Lévy, A.; Martsinovich, N. Adsorption Studies of P-Aminobenzoic Acid on the Anatase TiO2(101) Surface. Langmuir 2014, 30, 12306–12314. [Google Scholar] [CrossRef]
- Abbasi, A.; Abdelrasoul, A.; Sardroodi, J.J. Adsorption of CO and NO Molecules on Al, P and Si Embedded MoS2 Nanosheets Investigated by DFT Calculations. Adsorption 2019, 25, 1001–1017. [Google Scholar] [CrossRef]
- Kruczek, J.; Chiu, S.-W.; Varma, S.; Jakobsson, E.; Pandit, S.A. Interactions of Monovalent and Divalent Cations at Palmitoyl-Oleoyl-Phosphatidylcholine Interface. Langmuir 2019, 35, 10522–10532. [Google Scholar] [CrossRef] [PubMed]
- Dehghani, A.; Bahlakeh, G.; Ramezanzadeh, B.; Ramezanzadeh, M. Electronic/Atomic Level Fundamental Theoretical Evaluations Combined with Electrochemical/Surface Examinations of Tamarindus Indiaca Aqueous Extract as a New Green Inhibitor for Mild Steel in Acidic Solution (HCl 1 M). J. Taiwan Inst. Chem. Eng. 2019, 102, 349–377. [Google Scholar] [CrossRef]
- Shahini, M.H.; Keramatinia, M.; Ramezanzadeh, M.; Ramezanzadeh, B.; Bahlakeh, G. Combined Atomic-Scale/DFT-Theoretical Simulations & Electrochemical Assessments of the Chamomile Flower Extract as a Green Corrosion Inhibitor for Mild Steel in HCl Solution. J. Mol. Liq. 2021, 342, 117570. [Google Scholar] [CrossRef]
- Hammes-Schiffer, S. Quantum Effects in Complex Systems: Summarizing Remarks. Faraday Discuss. 2020, 221, 582–588. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | EHOMO | ELUMO | ∆E | IE | EA | Ƞ | χ | ω | σ | ∆N | ε | ω+ | ω− |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HTR | −6.22 | −2.116 | 4.104 | 6.22 | 2.116 | 2.052 | 4.168 | 4.232 | 0.487 | 0.158 | 0.236 | 0.545 | 4.713 |
| TRS | −6.733 | −6.355 | 0.378 | 6.733 | 6.355 | 0.189 | 6.544 | 113.29 | 5.291 | −4.560 | 0.008 | 53.420 | 59.964 |
| Molecule | Adsorption Energy (in eV) | |
|---|---|---|
| Aqueous Phase | Vacuum Phase | |
| HTR | −4.085 | −5.214 |
| TRS | −3.803 | −4.254 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lgaz, H.; Lee, H.-s. Computational Exploration of Phenolic Compounds in Corrosion Inhibition: A Case Study of Hydroxytyrosol and Tyrosol. Materials 2023, 16, 6159. https://doi.org/10.3390/ma16186159
Lgaz H, Lee H-s. Computational Exploration of Phenolic Compounds in Corrosion Inhibition: A Case Study of Hydroxytyrosol and Tyrosol. Materials. 2023; 16(18):6159. https://doi.org/10.3390/ma16186159
Chicago/Turabian StyleLgaz, Hassane, and Han-seung Lee. 2023. "Computational Exploration of Phenolic Compounds in Corrosion Inhibition: A Case Study of Hydroxytyrosol and Tyrosol" Materials 16, no. 18: 6159. https://doi.org/10.3390/ma16186159
APA StyleLgaz, H., & Lee, H.-s. (2023). Computational Exploration of Phenolic Compounds in Corrosion Inhibition: A Case Study of Hydroxytyrosol and Tyrosol. Materials, 16(18), 6159. https://doi.org/10.3390/ma16186159