
Long-Term Oxidation Susceptibility in Ambient Air of the Semiconductor Kesterite Cu2ZnSnS4 Nanopowders Made by Mechanochemical Synthesis Method



Abstract
:1. Introduction
2. Experimental
2.1. Preparation of Kesterite Nanopowder Materials
2.2. Sample Labeling
2.3. Characterization
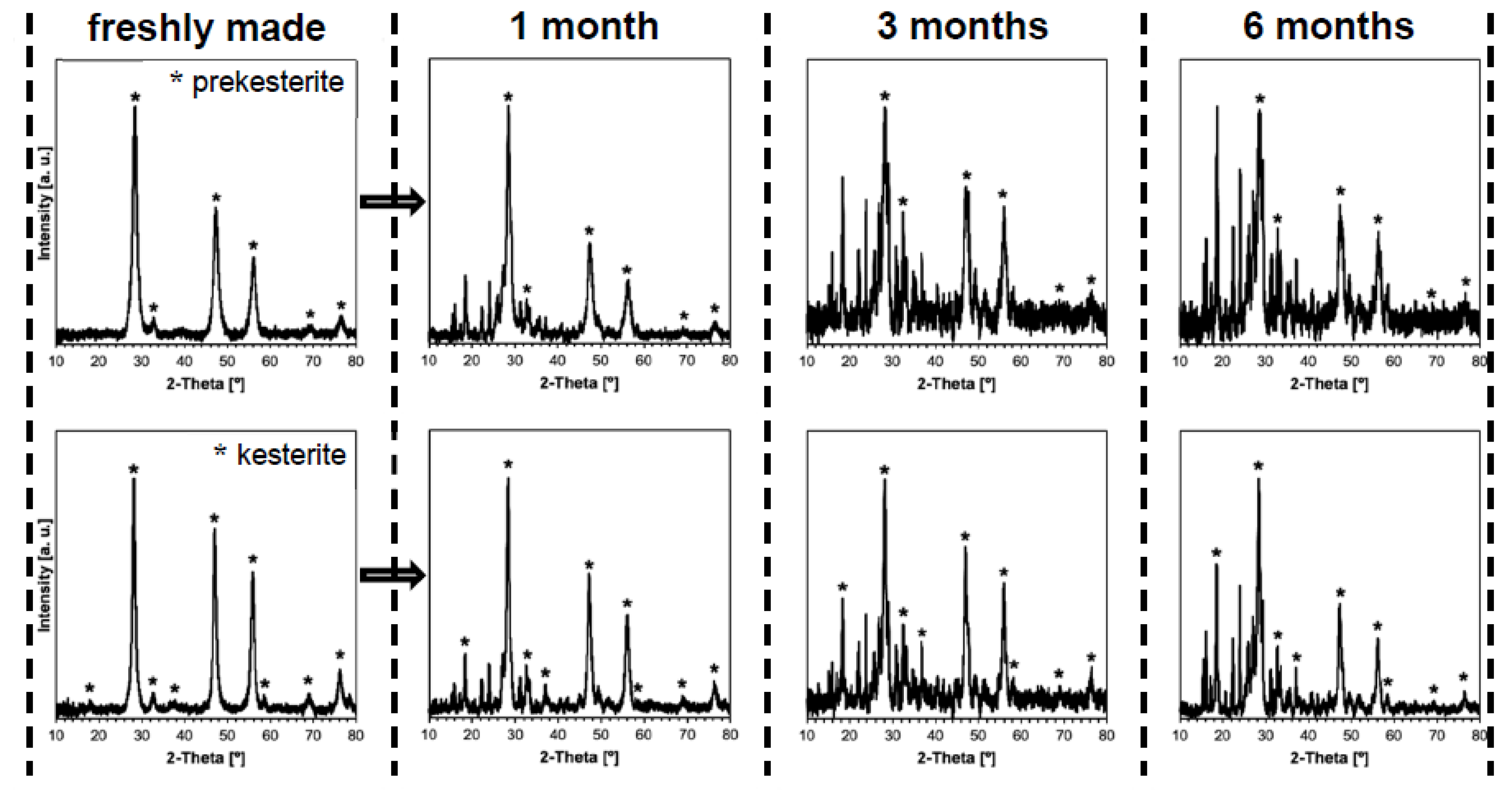
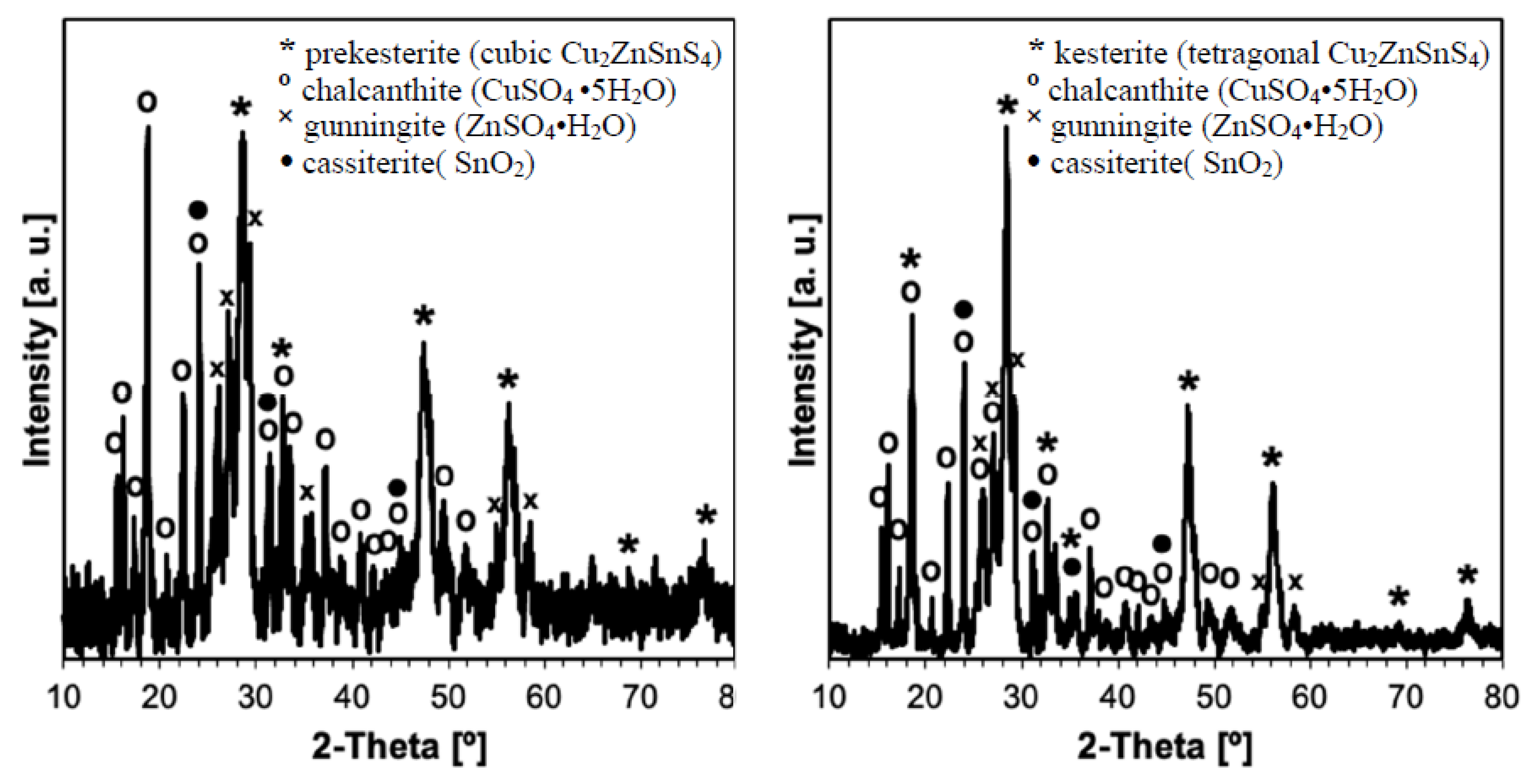
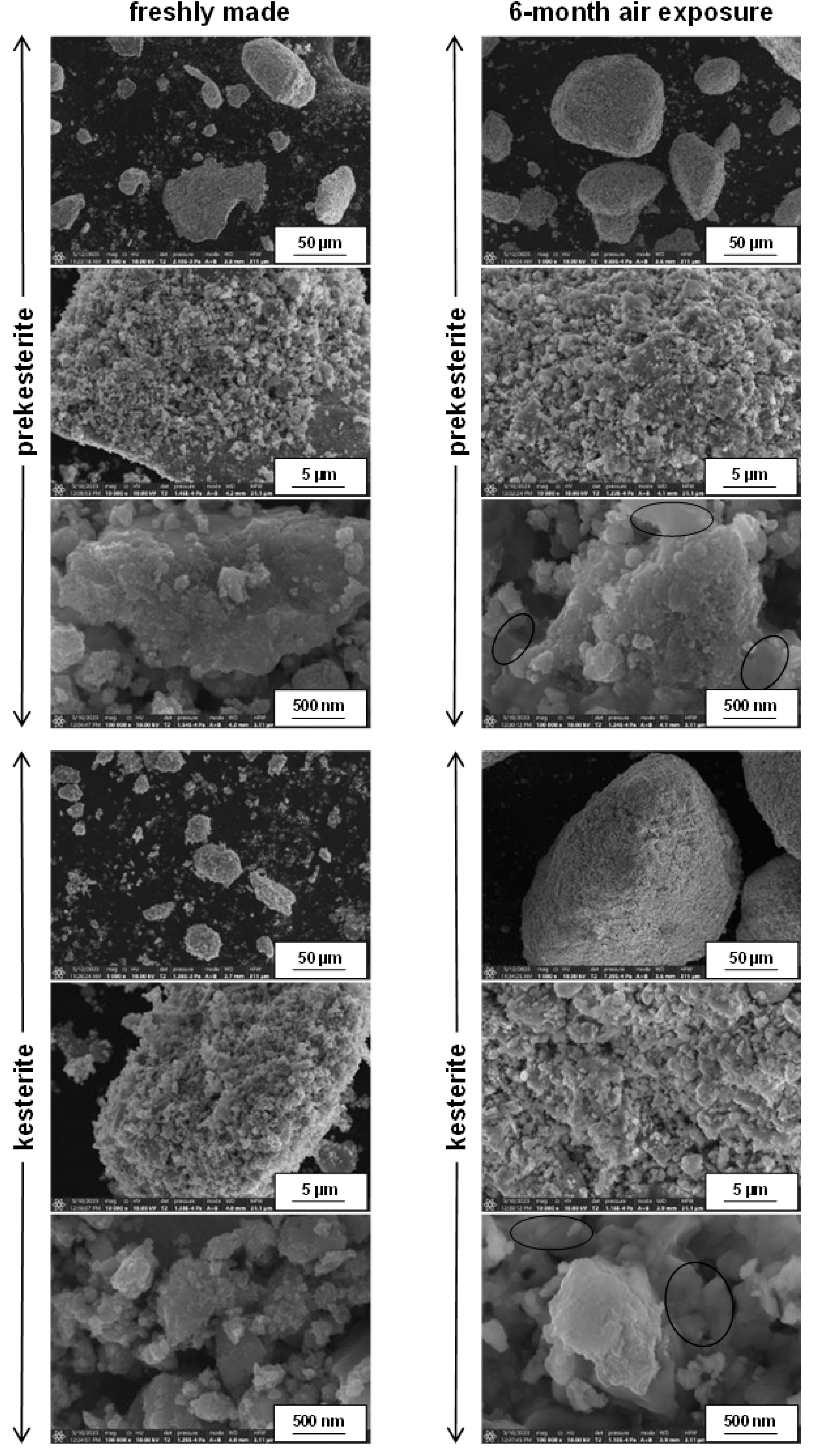
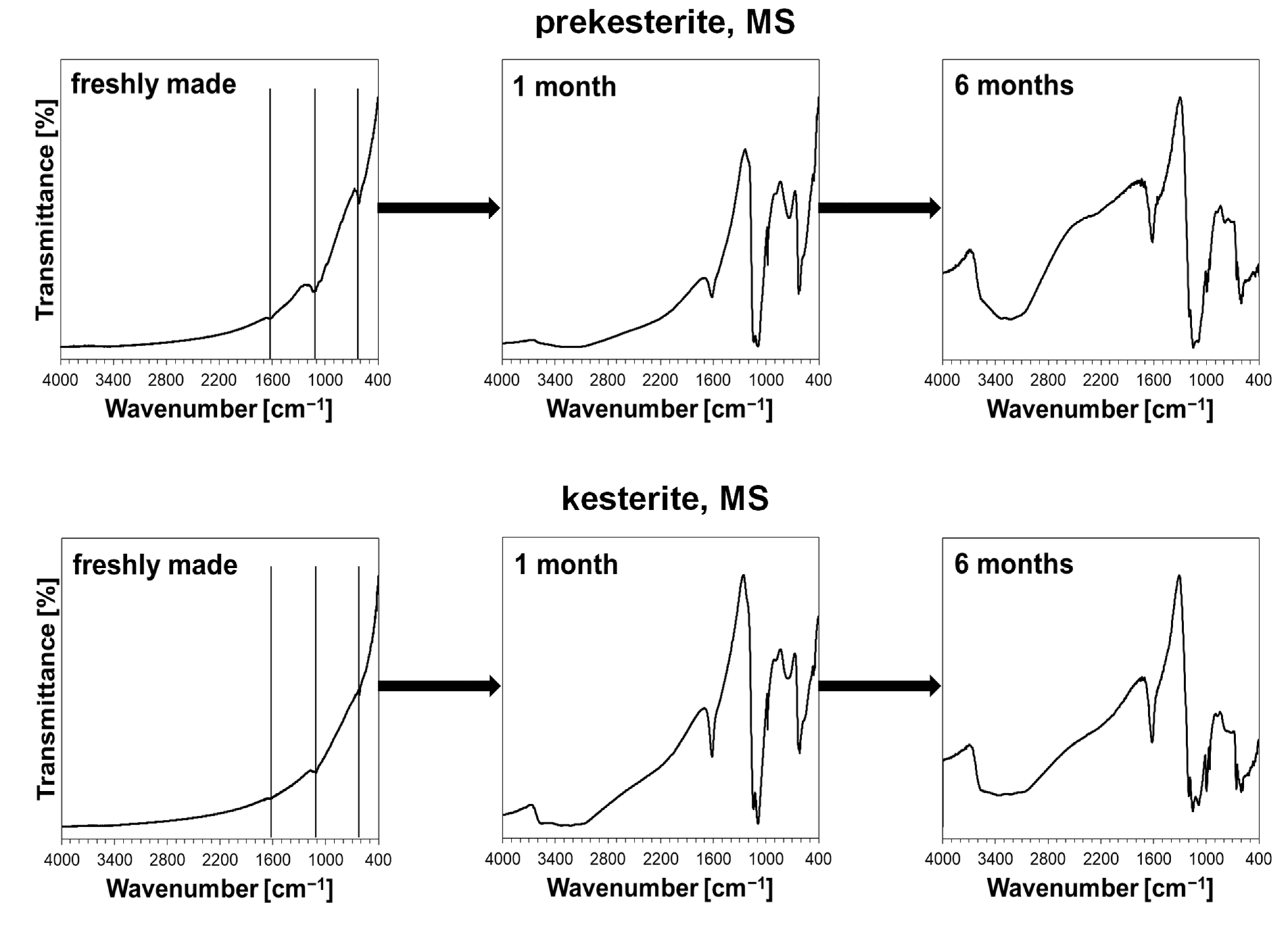
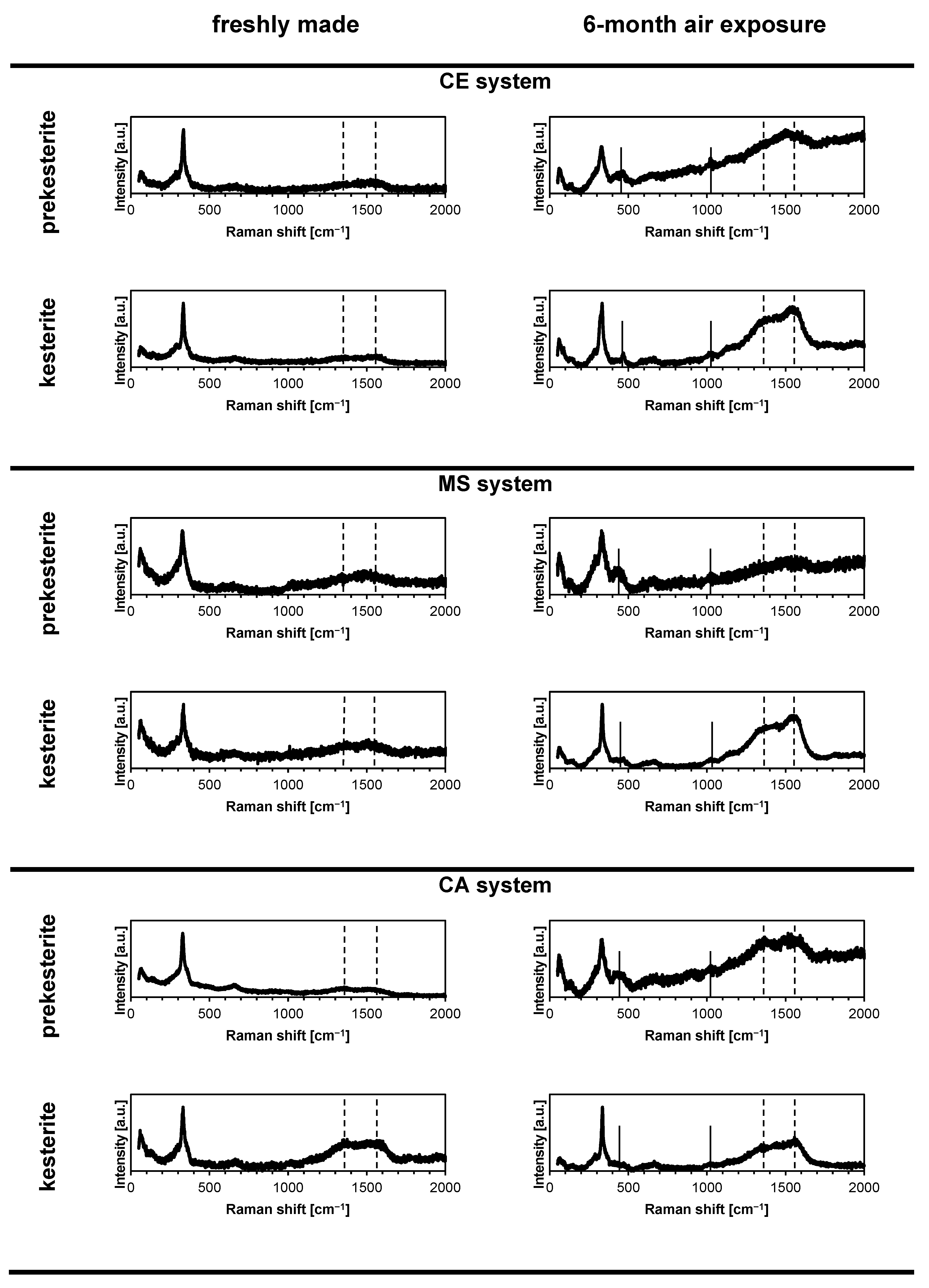
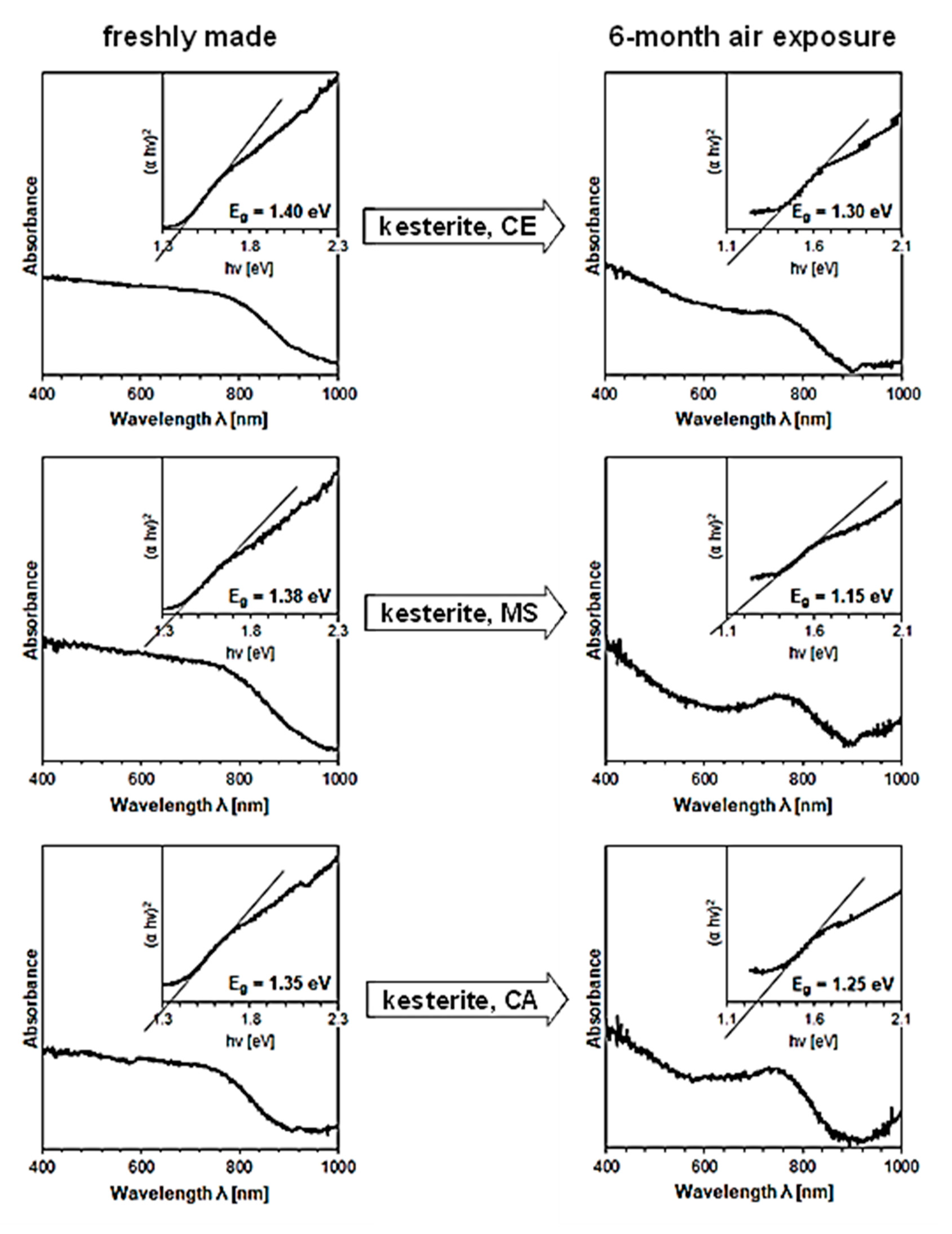
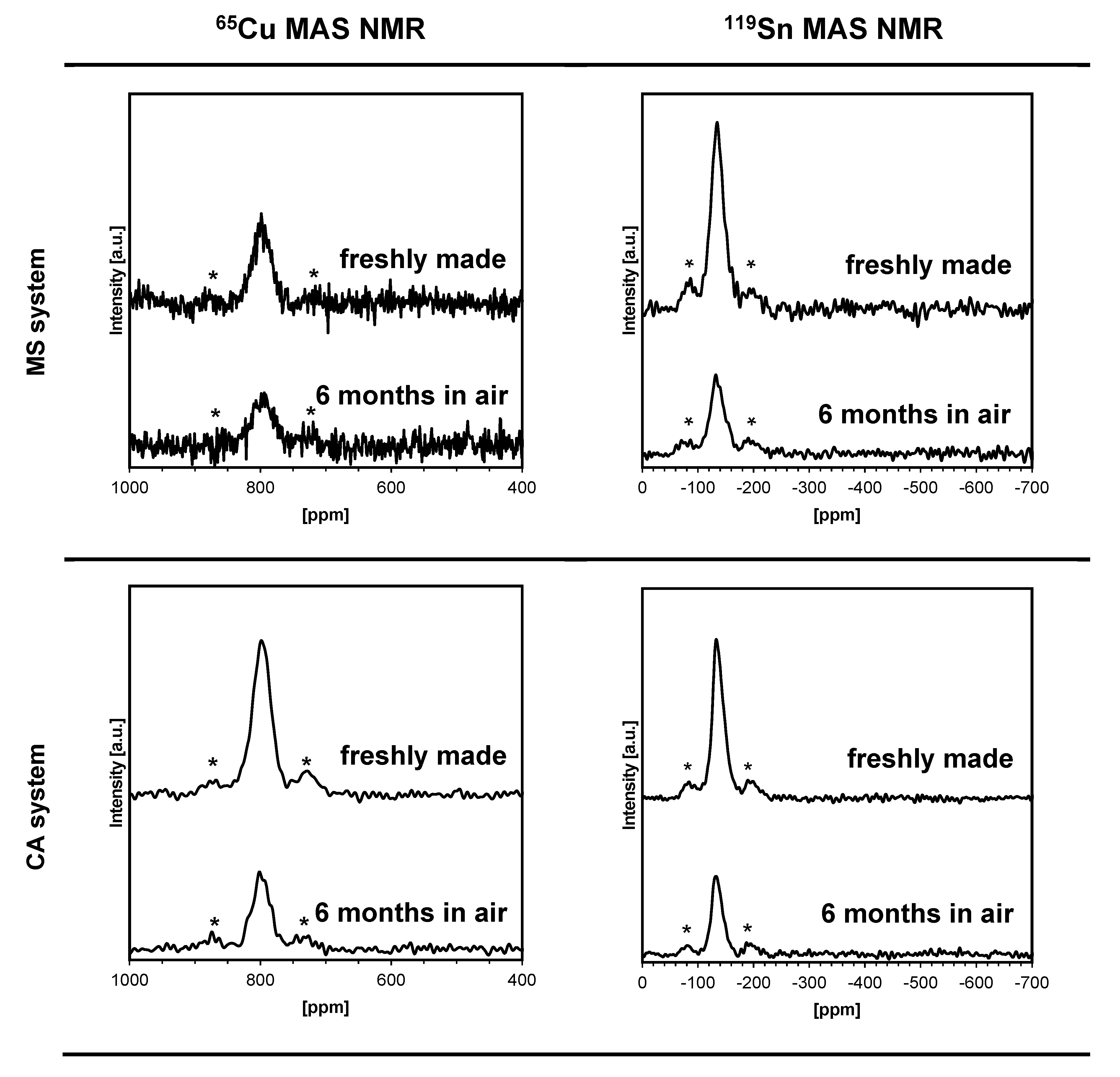
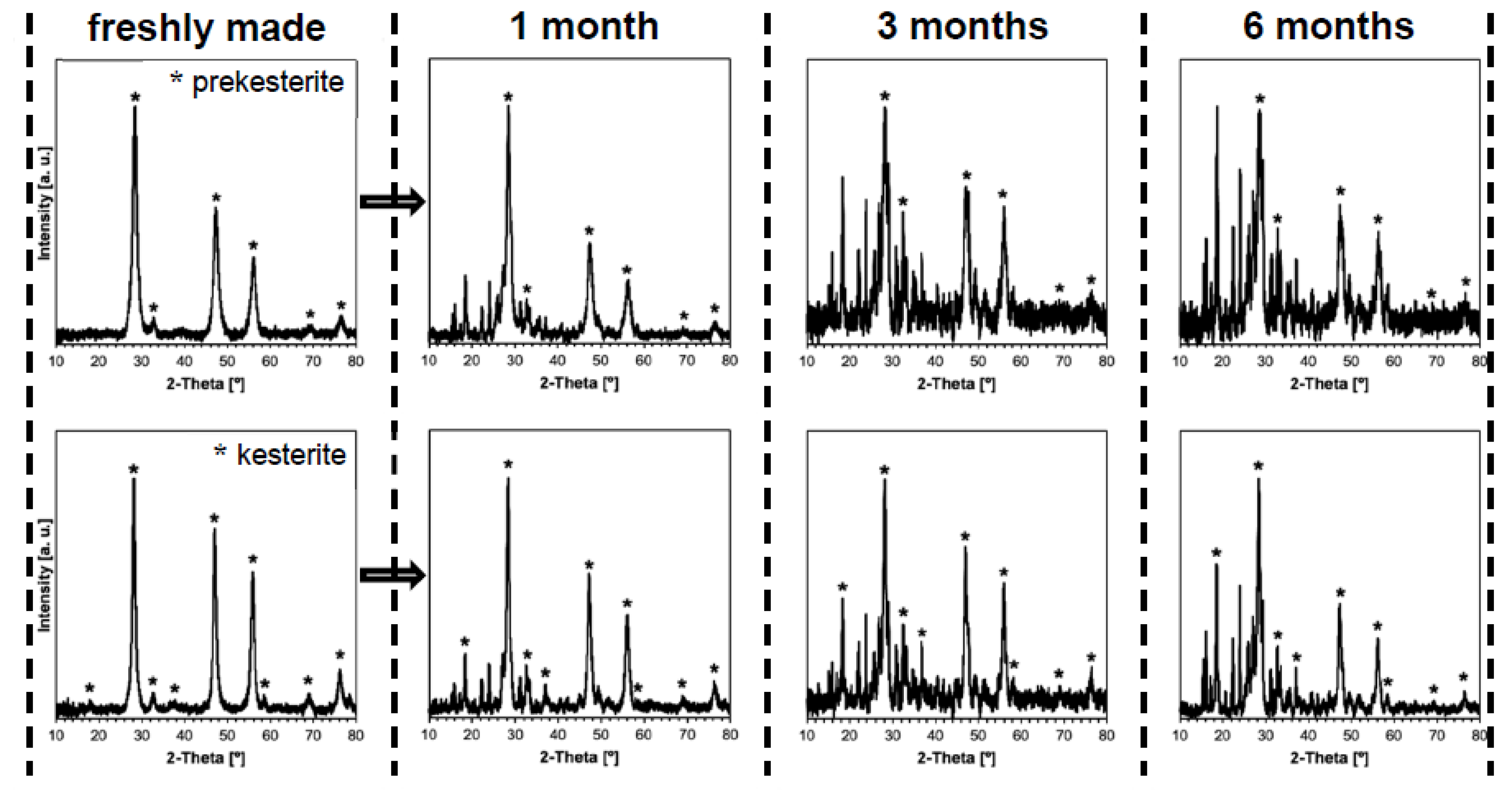
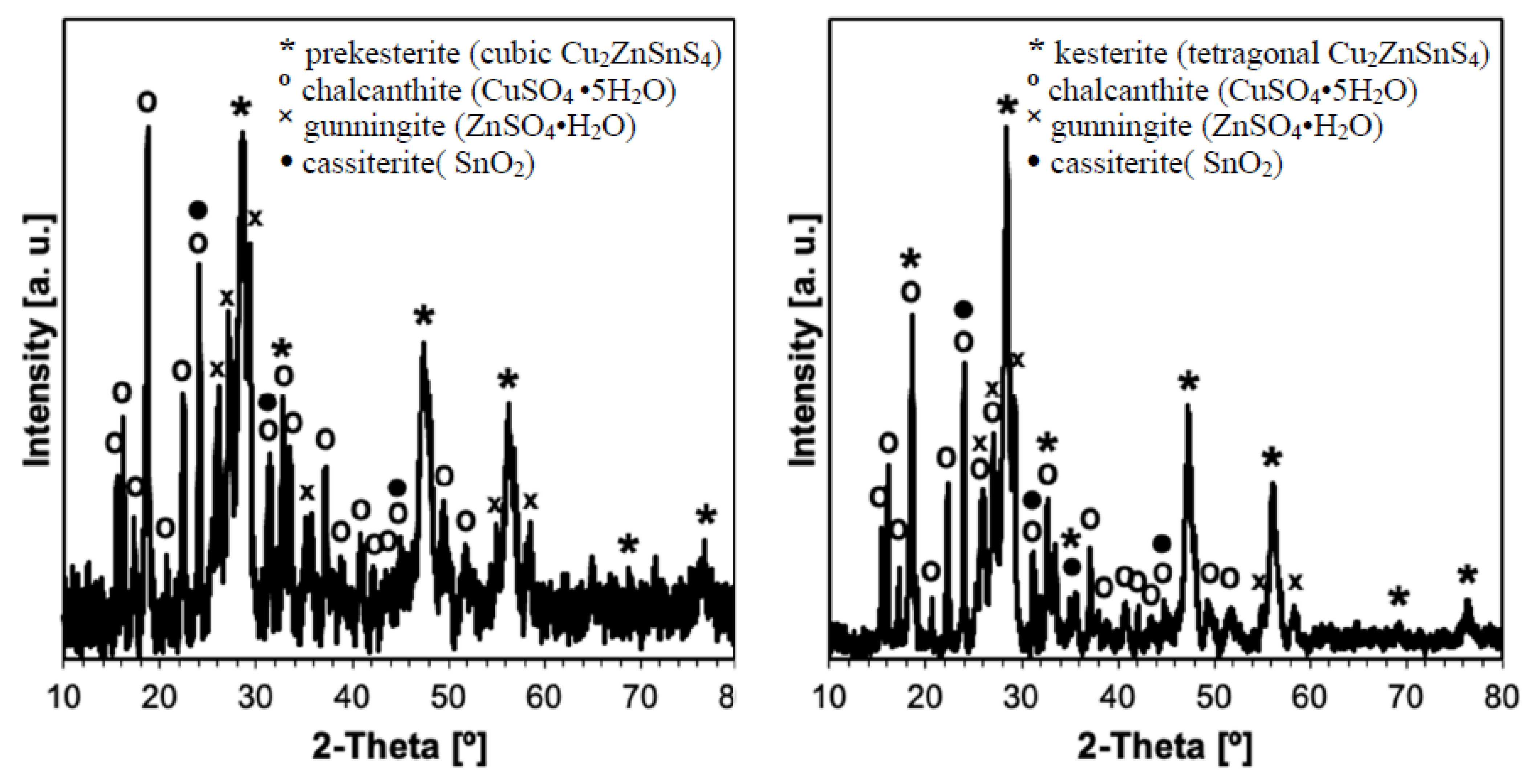
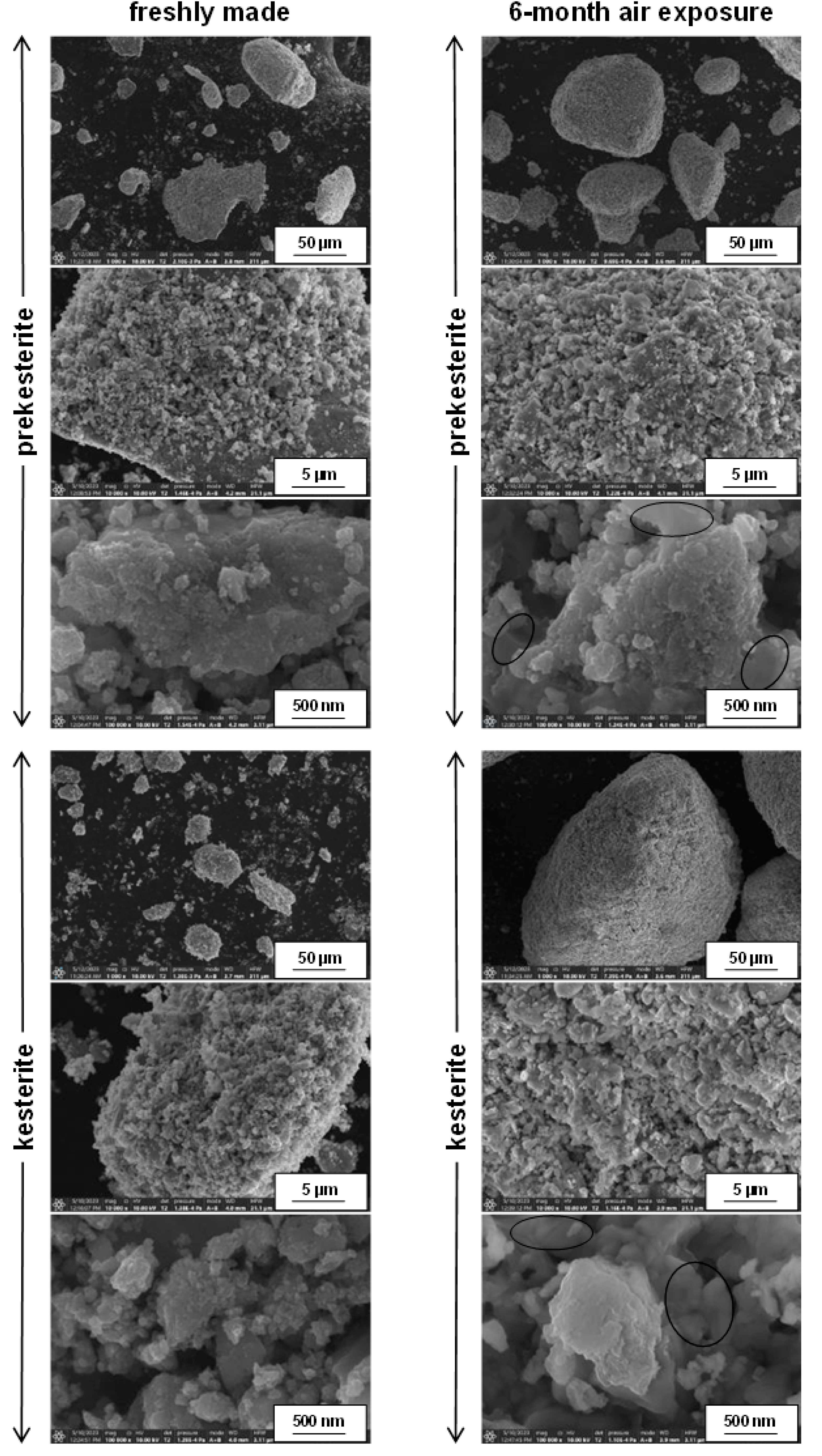
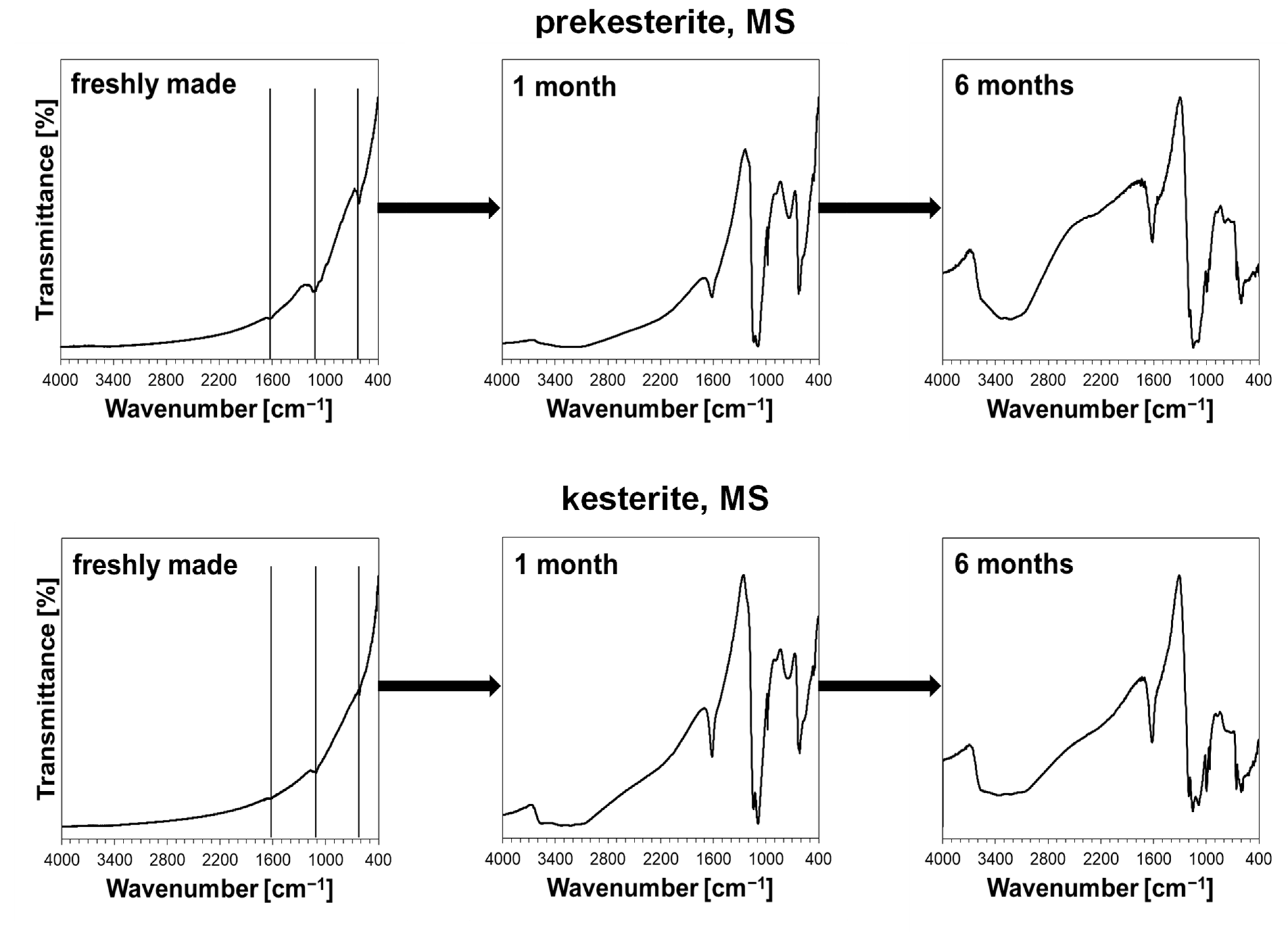
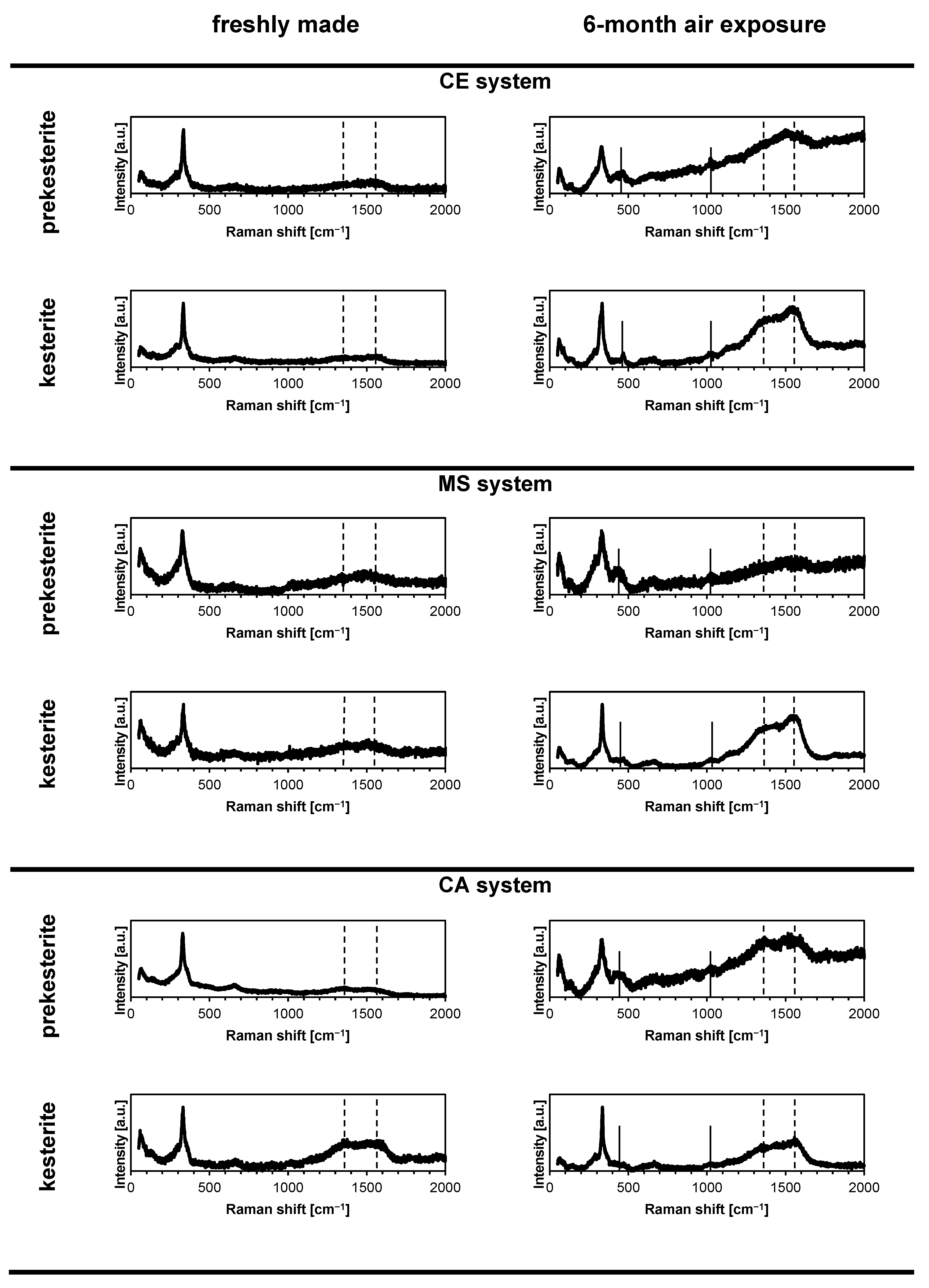
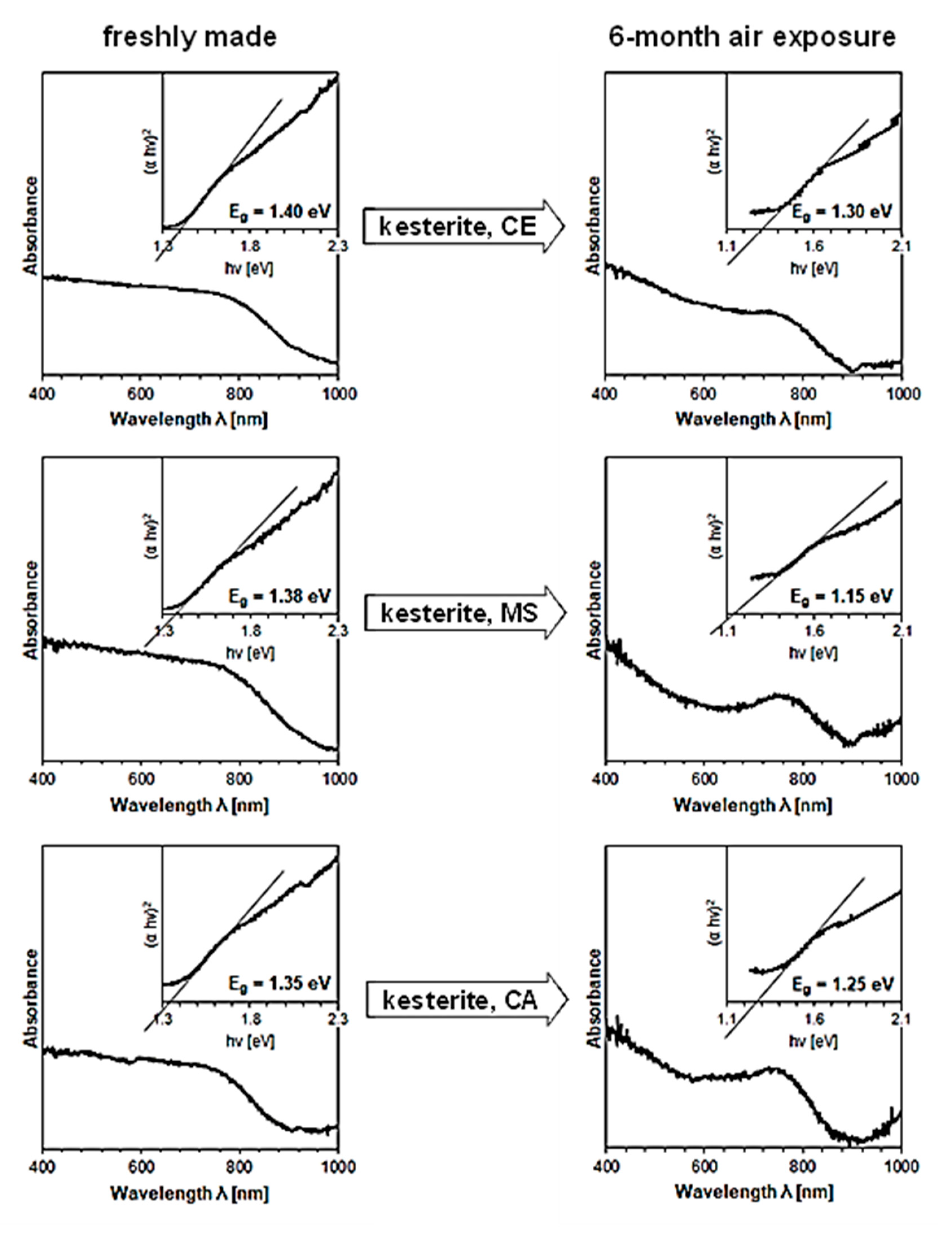
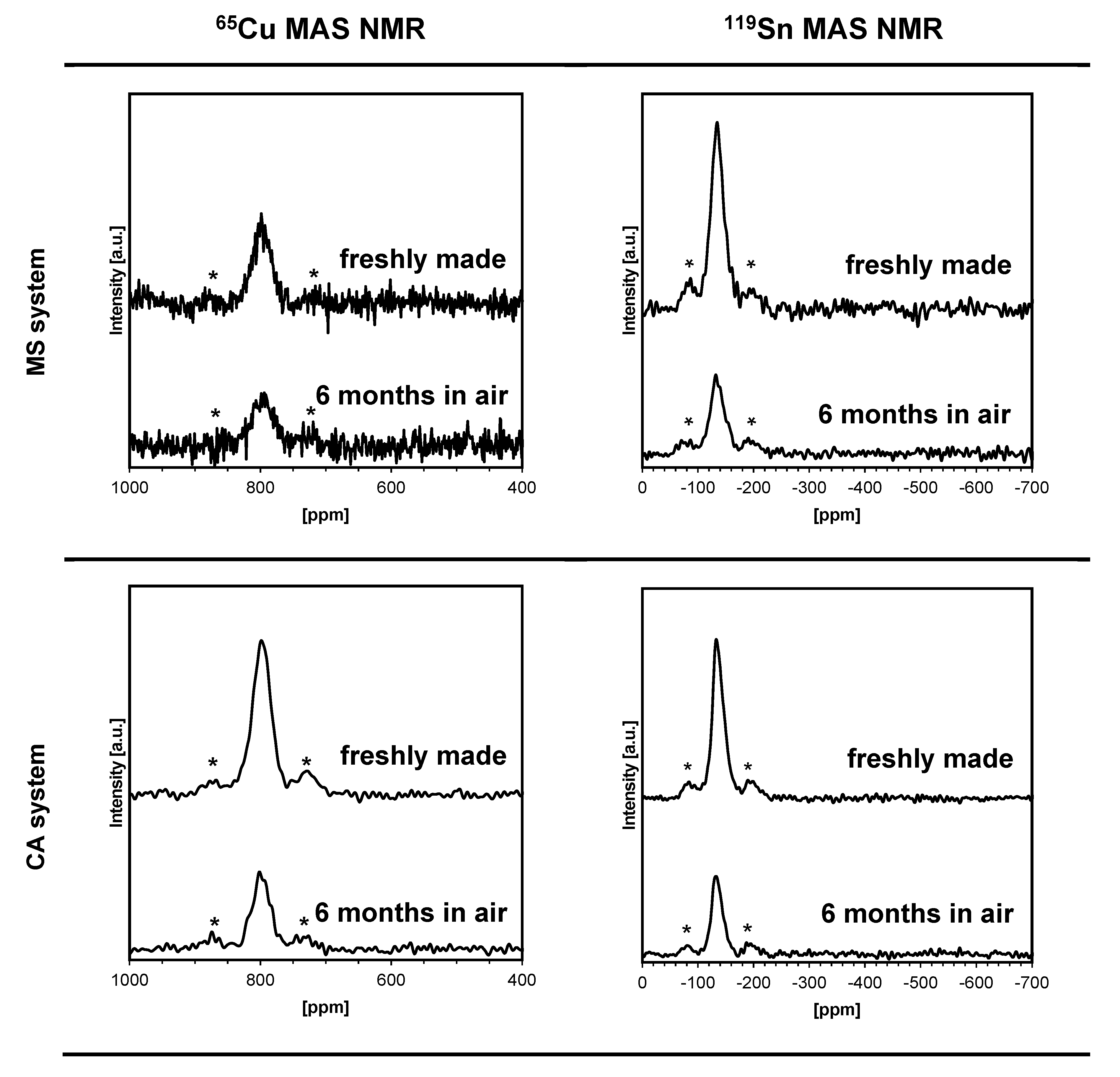
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wexler, R.B.; Gautam, G.S.; Carter, E.A. Optimizing kesterite solar cells from Cu2ZnSnS4 to Cu2CdGe(S,Se)4. J. Mater. Chem. A 2021, 9, 9882–9897. [Google Scholar] [CrossRef]
- Wallace, S.K.; Mitzi, D.B.; Walsh, A. The steady rise of kesterite solar cells. ACS Energy Lett. 2017, 2, 776–779. [Google Scholar] [CrossRef]
- Boerasu, J.; Vasile, B.S. Current status of the open-circuit voltage of kesterite CZTS absorber layers for photovoltaic applications—Part I, a Review. Materials 2022, 15, 8427. [Google Scholar] [CrossRef] [PubMed]
- Nazligul, A.S.; Wang, M.Q.; Choy, K.L. Recent development in earth-abundant kesterite materials and their applications. Sustainability 2020, 12, 5138. [Google Scholar] [CrossRef]
- Wibowo, R.A. Powder-to-film approach for fabricating critical raw material-free kesterite Cu2ZnSn(S,Se)4 thin film photovoltaic: A review. Sol. Energy 2018, 176, 157–169. [Google Scholar] [CrossRef]
- Ratz, T.; Brammertz, G.; Caballero, R.; León, M.; Canulescu, S.; Schou, J.; Gütay, L.; Pareek, D.; Taskesen, T.; Kim, D.H.; et al. Physical routes for the synthesis of kesterite. J. Phys. Energy 2019, 1, 042003. [Google Scholar] [CrossRef]
- Todorov, T.; Hillhouse, H.W.; Aazou, S.; Sekkat, Z.; Vigil-Galán, O.; Deshmukh, S.D.; Agrawal, R.; Bourdais, S.; Valdés, M.; Arnou, P. Solution-based synthesis of kesterite thin film semiconductors. J. Phys. Energy 2020, 2, 012003. [Google Scholar] [CrossRef]
- Podsiadlo, S.; Bialoglowski, M.; Matyszczak, G.; Marek, P.; Gebicki, W.; Bacewicz, R.; Stachowicz, M.; Dluzewski, P.; Wozniak, K. Synthesis of Bulk Kesterite—A Prospective Photovoltaic Material. Eur. J. Inorg. Chem. 2014, 2014, 4730–4733. [Google Scholar] [CrossRef]
- Sahu, M.; Reddy, V.S.M.; Kim, B.; Patro, B.; Park, C.; Kim, W.K.; Sharma, P. Fabrication of Cu2ZnSnS4 light absorber using a cost-effective mechanochemical method for photovoltaic applications. Materials 2022, 15, 1708. [Google Scholar] [CrossRef]
- Tan, D.; García, F. Main group mechanochemistry: From curiosity to established protocols. Chem. Soc. Rev. 2019, 48, 2274. [Google Scholar] [CrossRef]
- Kapusta, K.; Drygas, M.; Janik, J.F.; Jelen, P.; Bucko, M.M.; Olejniczak, Z. From magnetic cubic pre-kesterite to semiconducting tetragonal kesterite Cu2ZnSnS4 nanopowders via the mechano-chemically assisted route. J. Alloys Compd. 2019, 770, 981–988. [Google Scholar] [CrossRef]
- Lejda, K.; Drygaś, M.; Janik, J.F.; Szczytko, J.; Twardowski, A.; Olejniczak, Z. Magnetism of kesterite Cu2ZnSnS4 semiconductor nanopowders prepared by mechanochemically assisted synthesis method. Materials 2020, 13, 3487. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, K.; Drygas, M.; Janik, J.F.; Olejniczak, Z. New synthesis route to kesterite Cu2 ZnSnS4 semiconductor nanocrystalline powders utilizing copper alloys and a high energy ball milling-assisted process. J. Mater. Res. Technol. 2020, 9, 13320–13331. [Google Scholar] [CrossRef]
- Lejda, K.; Janik, J.F.; Perzanowski, M.; Stelmakh, S.; Pałosz, B. Oxygen aspects in the high-pressure and high-temperature sintering of semiconductor kesterite Cu2ZnSnS4 nanopowders prepared by a mechanochemically assisted synthesis method. Int. J. Mol. Sci. 2023, 24, 3159. [Google Scholar] [CrossRef] [PubMed]
- Dun, C.C.; Holzwarth, N.A.W.; Li, Y.; Huang, W.X.; Carroll, D.L. Cu2ZnSnSxO4−x and Cu2ZnSnSxSe4-x: First principles simulations of optimal alloy configurations and their energies. J. Appl. Phys. 2014, 115, 193513. [Google Scholar] [CrossRef]
- Tablero, C. Effect of the oxygen isoelectronic substitution in Cu2ZnSnS4 and its photovoltaic application. Thin Solid Film 2012, 520, 5011–5013. [Google Scholar] [CrossRef]
- Yu, R.S.; Hung, T.C. Influences of oxygen incorporation on the structural and optoelectronic properties of Cu2ZnSnS4 thin films. Appl. Surf. Sci. 2016, 364, 909–916. [Google Scholar] [CrossRef]
- Washio, T.; Shinji, T.; Tajima, S.; Fukano, T.; Motohiro, T.; Jimbo, K.; Katagiri, H. 6% efficiency Cu2ZnSnS4-based thin film solar cells using oxide precursors by open atmosphere type CVD. J. Mater. Chem. 2012, 22, 4021–4024. [Google Scholar] [CrossRef]
- Larsen, J.K.; Ren, Y.; Ross, N.; Sarhammer, E.; Li, S.Y.; Platzer-Bjorkman, C. Surface modification through air annealing Cu2ZnSn(S,Se)4 absorbers. Thin Solid Film 2017, 633, 118–121. [Google Scholar] [CrossRef]
- Tajima, S.; Asahi, R.; Isheim, D.; Seidman, D.N.; Itoh, T.; Hasegawa, M.; Ohishi, K. Atom-probe tomographic study of interfaces of Cu2ZnSnS4 photovoltaic cells. Appl. Phys. Lett. 2014, 105, 093901. [Google Scholar] [CrossRef]
- Hegedus, M.; Balaz, P.; Balaz, M.; Siffalovic, P.; Daneu, N.; Kanuchova, M.; Briancin, J.; Fabian, M. Mechanochemical approach to a Cu2ZnSnS4 solar cell absorber via a “micro-nano” route. J. Mater. Sci. 2018, 53, 13617–13630. [Google Scholar] [CrossRef]
- Havryliuk, Y.; Valakh, M.Y.; Dzhagan, V.; Greshchuk, O.; Yukhymchuk, V.; Raevskaya, A.; Stroyuk, O.; Selyshchev, O.; Gaponik, N.; Zahn, D.R.T. Raman characterization of Cu2ZnSnS4 nanocrystals: Phonon confinement effect and formation of CuxS phases. RSC Adv. 2018, 8, 30736–30746. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Nam, D.; Gansukh, M.; Park, S.-N.; Sung, S.-J.; Kim, D.-H.; Kang, J.-K.; Sai, C.D.; Tran, T.H.; Cheong, H. Influence of sulfate residue on Cu2ZnSnS4 thin films prepared by direct solution method. Sol. Energy Mater. Sol. Cells 2015, 136, 113–119. [Google Scholar] [CrossRef]
- Awadallah, O.; Cheng, Z. In situ Raman monitoring of Cu2ZnSnS4 oxidation and related decomposition at elevated temperature. IEEE J. Photovolt. 2016, 6, 764–769. [Google Scholar] [CrossRef]
- Ramakrishna, R.V.V.; Abraham, K.P. Kinetics of oxidation of copper sulfide. Metall. Trans. 1971, 2, 2463–2470. [Google Scholar] [CrossRef]
- ter Maat, H.; Hogendoorn, J.A.; Versteeg, G.F. The removal of hydrogen sulfide from gas streams using an aqueous metal sulfate absorbent. Part II. The regeneration of copper sulfide to copper oxide—An experimental study. Sep. Purif. Technol. 2005, 43, 199–213. [Google Scholar] [CrossRef]
- Todd, E.C.; Sherman, D.M. Surface oxidation of chalcocite (Cu2S) under aqueous (pH = 2–11) and ambient atmospheric conditions: Mineralogy from Cu L- and O K-edge X-ray absorption spectroscopy. Am. Mineral. 2003, 88, 1652–1656. [Google Scholar] [CrossRef]
- Baláž, M.; Dutková, E.; Bujňáková, Z.; Tóthová, E.; Kostova, N.G.; Karakirova, Y.; Briančin, J.; Kaňuchová, M. Mechanochemistry of copper sulfides: Characterization, surface oxidation and photocatalytic activity. J. Alloys Compd. 2018, 746, 576–582. [Google Scholar] [CrossRef]
- Reimers, G.W.; Hjelmstad, K.E. Analysis of the Oxidation of Chalcopyrite, Chalcocite, Galena, Pyrrhotite, Marcasite, and Arsenopyrite; Report of Investigations, 9118; U.S. Department of the Interior, Bureau of Mines: Pittsburgh, PA, USA, 1987.
- Mitovski, A.; Štrbac, N.; Sokić, M.; Kragović, M.; Grekulović, V. Reaction Mechanism and kinetics of sulfide copper concentrate oxidation at elevated temperatures. Metall. Mater. Eng. 2017, 23, 267–280. [Google Scholar] [CrossRef]
- Buehler, H.A.; Gottschalk, V.H. Oxidation of sulphides. Econ. Geol. 1910, 5, 28–35. [Google Scholar] [CrossRef]
- Steger, H.F.; Desjardins, L.E. Oxidation of sulfide minerals. V. Galena, sphalerite and chalcocite. Can. Mineral. 1980, 18, 365–372. [Google Scholar]
- Siriwardane, R.V.; Woodruff, S. In situ Fourier transform infrared characterization of sulfur species resulting from the reaction of water vapor and oxygen with zinc sulfide. Ind. Eng. Chem. Res. 1997, 36, 5277–5281. [Google Scholar] [CrossRef]
- Gulyaeva, R.I.; Selivanov, E.N.; Pikalov, S.M. Mechanism and kinetics of the thermal oxidation of natural sphalerite. Russ. Metall. (Met.) 2018, 3, 221–227. [Google Scholar] [CrossRef]
- Avellaneda, D.; Sánchez-Orozco, I.; Martínez, J.A.A.; Shaji, S.; Krishnan, B. Thin films of tin sulfides: Structure, composition and optoelectronic properties. Mater. Res. Express 2019, 6, 016409. [Google Scholar] [CrossRef]
- Hämmer, M.; Netzsch, P.; Steffen Klenner, S.; Neuschulz, K.; Struckmann, M.; Wickleder, M.S.; Daub, M.; Hillebrecht, H.; Pöttgen, R.; Höppe, H.A. The tin sulfates Sn(SO4)2 and Sn2(SO4)3: Crystal structures, optical and thermal properties. Dalton Trans. 2021, 50, 12913–12922. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.A.K.; Fjellvåg, H.; Kjekshus, A. Synthesis and characterization of tis sulfates and oxide sulfate. Acta Chem. Scand. 1998, 52, 305–311. [Google Scholar] [CrossRef]
- Edwards, R.; Gillard, R.D.; Williams, P.A. The stabilities of secondary tin minerals. Part 2*. The hydrolysis of tin(II) sulphate and the stability of Sn3O(OH)2SO4. Mineral. Mag. 1996, 60, 427–432. [Google Scholar] [CrossRef]
- Posnjak, E. The nature of stannic acids. J. Phys. Chem. 1926, 30, 1073–1077. [Google Scholar] [CrossRef]
- Lee, J.I.; Lee, B.S.; Lee, J.Y.; Shin, J.Y.; Kim, T.W.; Ryu, J.H. A simple route for synthesis of SnO2 from copper alloy dross. J. Korean Cryst. Growth Cryst. Technol. 2014, 24, 84–87. [Google Scholar] [CrossRef]
- Dante, R.; Sliepcevich, A.; Andreoni, M.; Cotilli, M. Interference between tin sulfides, graphite and novolak oxidation. SAE Int. J. Mater. Manuf. 2018, 11, 89–94. [Google Scholar] [CrossRef]
- Gamo, I. Infrared absorption spectra of water of crystallization in copper sulfate penta- and monohydrate crystals. Bull. Chem. Soc. Jpn. 1961, 34, 764–766. [Google Scholar] [CrossRef]
- Spectrabase.com. Available online: https://spectrabase.com/spectrum/6L0EWWiHsDw (accessed on 10 August 2023).
- Akram, M.; Saleh, A.T.; Ibrahim, W.A.W.; Awan, A.S.; Hussain, R. Continuous microwave flow synthesis (CMFS) of nano-sized tin oxide: Effect of precursor concentration. Ceram. Int. 2016, 42, 8613–8619. [Google Scholar] [CrossRef]
- Fu, X.J.; Yang, G.; Sun, J.B.; Zhou, J. Vibrational spectra of copper sulfate hydrates investigated with low-temperature Raman spectroscopy and terahertz time domain spectroscopy. J. Phys. Chem. A 2012, 116, 7314–7318. [Google Scholar] [CrossRef] [PubMed]
- Apopei, A.I.; Buzgar, N. The Raman study of weathering minerals from the Coranda-Hondol open pit (Certej gold-silver deposit) and their photochemical degradation products under laser irradiation. Can. Mineral. 2014, 52, 1027–1038. [Google Scholar] [CrossRef]
- Bokobza, L.; Bruneel, J.L.; Couzi, M. Raman spectra of carbon-based materials (from graphite to carbon black) and of some silicone composites. C—J. Carbon Res. 2015, 1, 77–94. [Google Scholar] [CrossRef]
- Abdullahi, S.S.; Güner, S.; Koseoglu, Y.; Musa, I.M.; Adamu, B.I.; Abdulhamid, M.I. Sımple method for the determınatıon of band gap of a nanopowdered sample usıng Kubelka Munk theory. J. Niger. Assoc. Math. Phys. 2016, 35, 241–246. [Google Scholar]
- Choubrac, L.; Paris, M.; Lafond, A.; Guillot-Deudon, C.; Rocquefelte, X.; Jobic, S. Multinuclear (67Zn, 119Sn and 65Cu) NMR spectroscopy—An ideal technique to probe the cationic ordering in Cu2ZnSnS4 photovoltaic materials. Phys. Chem. Chem. Phys. 2013, 15, 10722–10725. [Google Scholar] [CrossRef]
- WWW-MINCRYST, Crystallographic and Crystallochemical Database for Minerals and Their Structural Analogues. 2018. Available online: http://database.iem.ac.ru/mincryst (accessed on 2 August 2023).
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The determination of pore volume and area distributions in porous substances. I. Computations from nitrogen isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cubic Zincblende-Type Prekesterite | Disordered Tetragonal Kesterite | |||||
|---|---|---|---|---|---|---|
| a [Å] | Dav [nm] | wt% | a/c [Å] | Dav [nm] | wt% | |
| CE system | ||||||
| freshly made | 5.44 | 6 | 100 | 5.43/10.86 | 12 | 100 |
| 1 month | 5.42 | 12 | 70 | 5.43/10.96 | 12 | 66 |
| 3 month | 5.45 | 9 | 42 | 5.45/10.98 | 12 | 46 |
| 6 month | 5.42 | 11 | 39 | 5.42/10.88 | 13 | 44 |
| MS system | ||||||
| freshly made | 5.42 | 7 | 100 | 5.43/10.83 | 13 | 100 |
| 1 month | 5.43 | 9 | 62 | 5.42/10.94 | 13 | 65 |
| 3 month | 5.44 | 12 | 43 | 5.44/10.98 | 12 | 57 |
| 6 month | 5.41 | 9 | 38 | 5.44/10.93 | 12 | 50 |
| CA system | ||||||
| freshly made | 5.43 | 9 | 100 | 5.44/10.82 | 18 | 100 |
| 1 month | 5.42 | 9 | 55 | 5.42/10.90 | 14 | 66 |
| 3 month | 5.44 | 9 | 36 | 5.44/10.99 | 15 | 65 |
| 6 month | 5.41 | 11 | 31 | 5.43/10.93 | 14 | 52 |
| CE System | MS System | CA System | ||||
|---|---|---|---|---|---|---|
| Prekesterite Kesterite | Prekesterite Kesterite | Prekesterite Kesterite | ||||
| HELIUM DENSITY dHe [g/cm3] | ||||||
| freshly made | 3.64 | 3.91 | 3.90 | 4.24 | 3.6 | 4.3 |
| 6-month exposure | 3.12 | 3.25 | 2.86 | 3.25 | 3.04 | 3.51 |
| BET/BJH SPECIFIC SURFACE AREA [m2/g] | ||||||
| freshly made | 18.2/21.9 | 17.3/21.1 | 17.9/21.9 | 17.2/21.1 | 12.7/13.0 | 11.4/11.6 |
| 6-month exposure | 12.0/12.8 | 21.8/25.9 | 10.6/11.7 | 21.1/24.2 | 11.4/11.8 | 19.5/21.9 |
| CE System | MS System | CA System | ||||
|---|---|---|---|---|---|---|
| Prekesterite | Kesterite | Prekesterite | Kesterite | Prekesterite | Kesterite | |
| OXYGEN CONTENT [wt%] | ||||||
| freshly made | 4.48 | 0.63 | 5.86 | 1.16 | 4.32 | 1.60 |
| 1 month | 22.4 | 25.4 | 22.1 | 24.7 | 28.2 | 24.8 |
| 3 month | 28.7 | 31.4 | 31.2 | 33.3 | 32.2 | 33.4 |
| 6 month | 31.8 | 36.3 | 33.3 | 32.8 | 33.6 | 34.0 |
| HYDROGEN CONTENT [wt%] | ||||||
| freshly made | 0.30 | 0.01 | 0.32 | 0.02 | 0.46 | 0.04 |
| 1 month | 2.04 | 2.26 | 1.88 | 2.29 | 2.38 | 2.20 |
| 3 month | 2.38 | 2.44 | 2.38 | 2.37 | 2.58 | 2.30 |
| 6 month | 3.00 | 2.79 | 3.06 | 2.78 | 3.04 | 2.67 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lejda, K.; Ziąbka, M.; Olejniczak, Z.; Janik, J.F. Long-Term Oxidation Susceptibility in Ambient Air of the Semiconductor Kesterite Cu2ZnSnS4 Nanopowders Made by Mechanochemical Synthesis Method. Materials 2023, 16, 6160. https://doi.org/10.3390/ma16186160
Lejda K, Ziąbka M, Olejniczak Z, Janik JF. Long-Term Oxidation Susceptibility in Ambient Air of the Semiconductor Kesterite Cu2ZnSnS4 Nanopowders Made by Mechanochemical Synthesis Method. Materials. 2023; 16(18):6160. https://doi.org/10.3390/ma16186160
Chicago/Turabian StyleLejda, Katarzyna, Magdalena Ziąbka, Zbigniew Olejniczak, and Jerzy Franciszek Janik. 2023. "Long-Term Oxidation Susceptibility in Ambient Air of the Semiconductor Kesterite Cu2ZnSnS4 Nanopowders Made by Mechanochemical Synthesis Method" Materials 16, no. 18: 6160. https://doi.org/10.3390/ma16186160
APA StyleLejda, K., Ziąbka, M., Olejniczak, Z., & Janik, J. F. (2023). Long-Term Oxidation Susceptibility in Ambient Air of the Semiconductor Kesterite Cu2ZnSnS4 Nanopowders Made by Mechanochemical Synthesis Method. Materials, 16(18), 6160. https://doi.org/10.3390/ma16186160