
Influence of Drying Technique on Physicochemical Properties of Synthetic Hydroxyapatite and Its Potential Use as a Drug Carrier
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Preparation of Hydroxyapatite
2.3. Determination of Density
- m1—mass of empty pycnometer (g),
- m2—mass of pycnometer with powder (g),
- m3—mass of pycnometer filled with water (g),
- m4—mass of pycnometer with powder filled with water (g),
- d0—density of water at measurement temperature (g/cm3),
- drz—actual density of hydroxyapatite (g/cm3).
2.4. X-ray Diffraction Analysis
2.5. Fourier-Transform Infrared Spectroscopy Analysis
2.6. Determination of Average Grain Diameter
2.7. Synthesis of Clindamycin-Modified Hydroxyapatite
2.8. Kinetic Release Studies of Antibiotic
2.9. Morphology Analysis
3. Results
3.1. Determination of Density
3.2. X-ray Diffraction Analysis
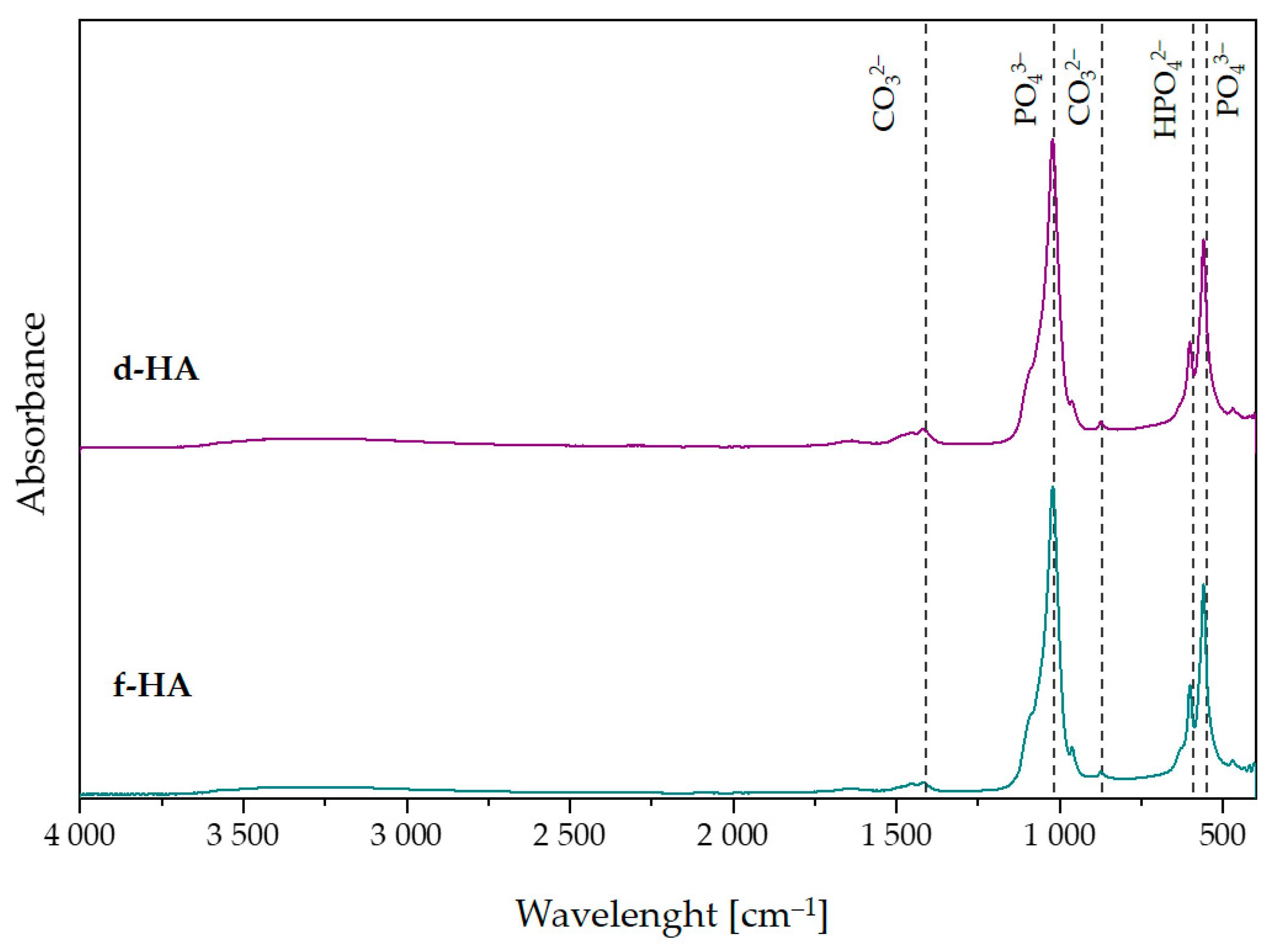
3.3. Fourier-Transform Infrared Spectroscopy Analysis
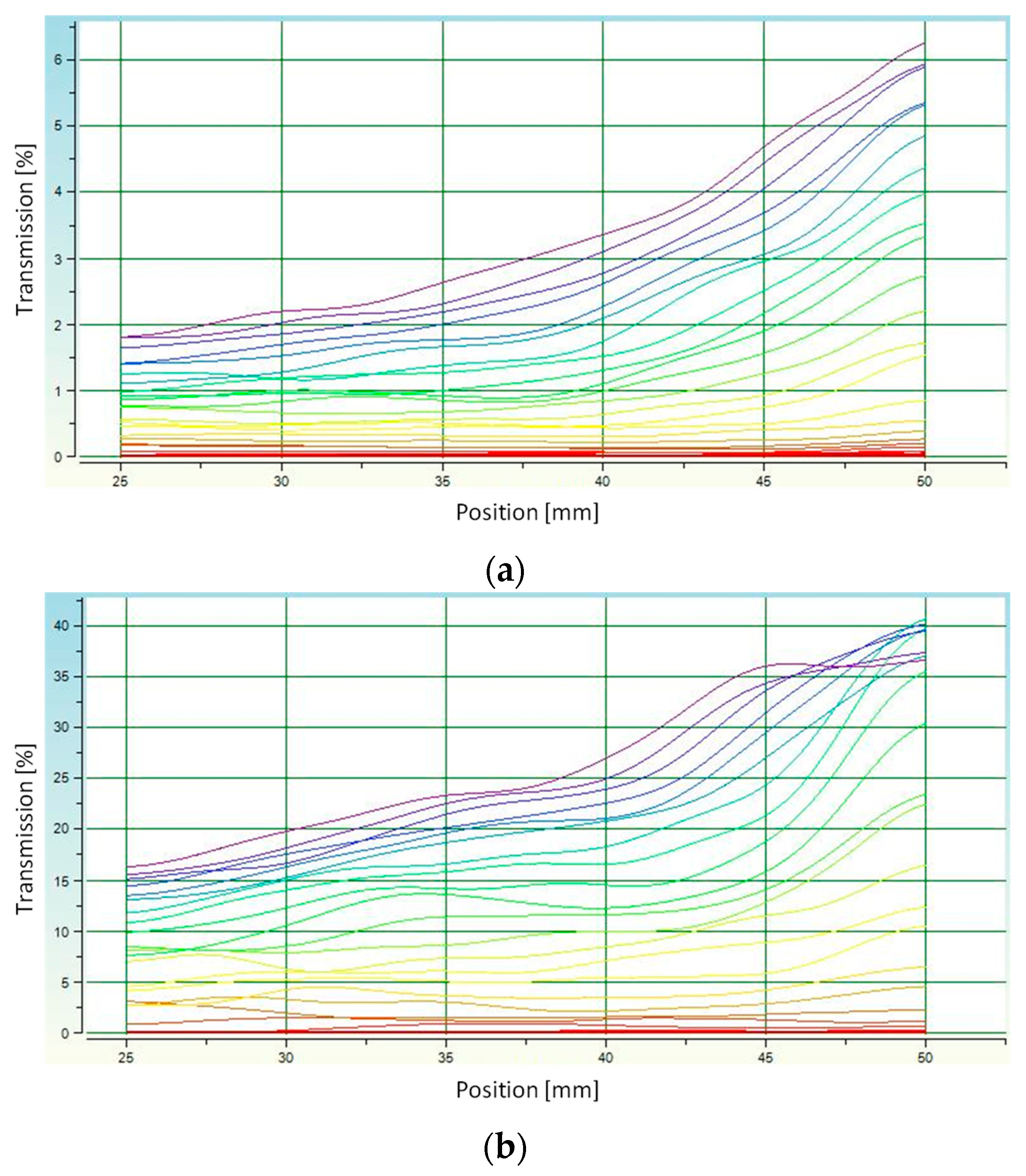
3.4. Determination of Average Grain Diameter
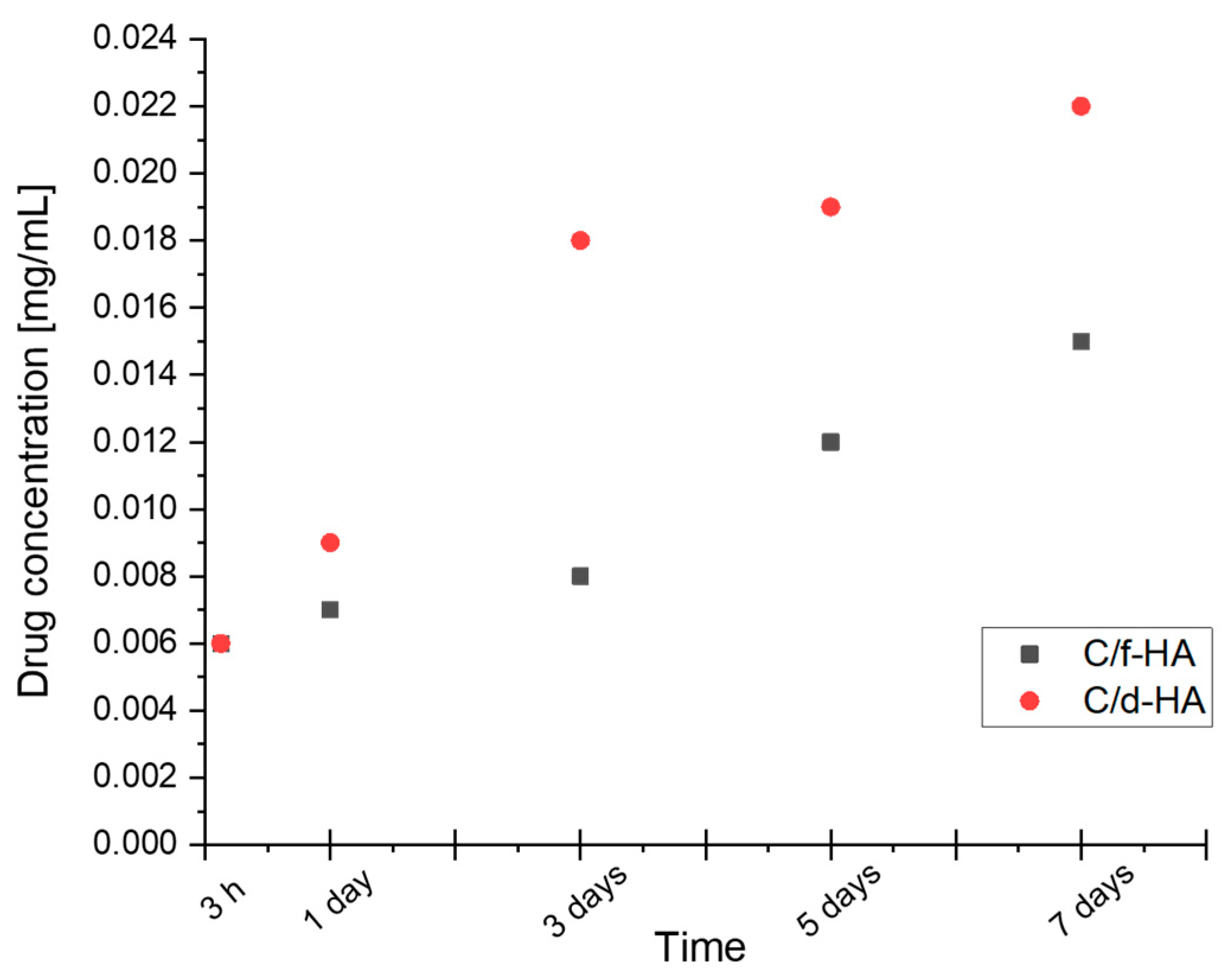
3.5. Kinetic Release Studies of Antibiotic
3.6. Morphology Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ezike, T.C.; Okpala, U.S.; Onoja, U.L.; Nwike, C.P.; Ezeako, E.C.; Okpara, O.J.; Okoroafor, C.C.; Eze, S.C.; Kalu, O.L.; Odoh, E.C.; et al. Advances in Drug Delivery Systems, Challenges and Future Directions. Heliyon 2023, 9, e17488. [Google Scholar] [CrossRef]
- Shivakalyani Adepu, S.R. Controlled Drug Delivery Systems: Current Status and Future Directions. Molecules 2021, 26, 5905. [Google Scholar] [CrossRef]
- Ghezzi, M.; Pescina, S.; Padula, C.; Santi, P.; Del Favero, E.; Cantù, L.; Nicoli, S. Polymeric Micelles in Drug Delivery: An Insight of the Techniques for Their Characterization and Assessment in Biorelevant Conditions. J. Control. Release 2021, 332, 312–336. [Google Scholar] [CrossRef] [PubMed]
- Majumder, N.; Das, N.G.; Das, S.K. Polymeric Micelles for Anticancer Drug Delivery. Ther. Deliv. 2020, 11, 613–635. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, E.; Yang, J.; Cao, Z. Strategies to Improve Micelle Stability for Drug Delivery. Nano Res. 2018, 11, 4985–4998. [Google Scholar] [CrossRef] [PubMed]
- Farooque, F.; Wasi, M.; Mughees, M.M. Liposomes as Drug Delivery System: An Updated Review. J. Drug Deliv. Ther. 2021, 11, 149–158. [Google Scholar] [CrossRef]
- Guimarães, D.; Cavaco-Paulo, A.; Nogueira, E. Design of Liposomes as Drug Delivery System for Therapeutic Applications. Int. J. Pharm. 2021, 601, 120571. [Google Scholar] [CrossRef]
- Zhang, W.Y.; Du, F.; He, M.; Bai, L.; Gu, Y.Y.; Yang, L.L.; Liu, Y.J. Studies of Anticancer Activity in Vitro and in Vivo of Iridium(III) Polypyridyl Complexes-Loaded Liposomes as Drug Delivery System. Eur. J. Med. Chem. 2019, 178, 390–400. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, C.; Pang, Z. Dendrimer-Based Drug Delivery Systems for Brain Targeting. Biomolecules 2019, 9, 790. [Google Scholar] [CrossRef]
- Wang, J.; Li, B.; Qiu, L.; Qiao, X.; Yang, H. Dendrimer-Based Drug Delivery Systems: History, Challenges, and Latest Developments. J. Biol. Eng. 2022, 16, 18. [Google Scholar] [CrossRef]
- Chauhan, A.S. Dendrimers for Drug Delivery. Molecules 2018, 23, 938. [Google Scholar] [CrossRef] [PubMed]
- Gelperina, S.; Kisich, K.; Iseman, M.D.; Heifets, L. The Potential Advantages of Nanoparticle Drug Delivery Systems in Chemotherapy of Tuberculosis. Am. J. Respir. Crit. Care Med. 2005, 172, 1487–1490. [Google Scholar] [CrossRef] [PubMed]
- Prakasam, M.; Locs, J.; Salma-Ancane, K.; Loca, D.; Largeteau, A.; Berzina-Cimdina, L. Fabrication, Properties and Applications of Dense Hydroxyapatite: A Review. J. Funct. Biomater. 2015, 6, 1099–1140. [Google Scholar] [CrossRef]
- Akpan, E.S.; Dauda, M.; Kuburi, L.S.; Obada, D.O.; Bansod, N.D.; Dodoo-Arhin, D. Hydroxyapatite Ceramics Prepared from Two Natural Sources by Direct Thermal Conversion: From Material Processing to Mechanical Measurements. Mater. Today Proc. 2020, 38, 2291–2294. [Google Scholar] [CrossRef]
- Ma, G. Three Common Preparation Methods of Hydroxyapatite. IOP Conf. Ser. Mater. Sci. Eng. 2019, 688, 033057. [Google Scholar] [CrossRef]
- Benedini, L.; Placente, D.; Ruso, J.; Messina, P. Adsorption/Desorption Study of Antibiotic and Anti-Inflammatory Drugs onto Bioactive Hydroxyapatite Nano-Rods. Mater. Sci. Eng. C 2019, 99, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; He, X.; Wu, Z. Mesoporous Hydroxyapatite: Preparation, Drug Adsorption, and Release Properties. Mater. Chem. Phys. 2014, 148, 153–158. [Google Scholar] [CrossRef]
- Moriguchi, Y.; Lee, D.S.; Chijimatsu, R.; Thamina, K.; Masuda, K.; Itsuki, D.; Yoshikawa, H.; Hamaguchi, S.; Myoui, A. Impact of Non-Thermal Plasma Surface Modification on Porous Calcium Hydroxyapatite Ceramics for Bone Regeneration. PLoS ONE 2018, 13, e0194303. [Google Scholar] [CrossRef] [PubMed]
- Benjumeda Wijnhoven, I.; Vallejos, R.; Santibanez, J.F.; Millán, C.; Vivanco, J.F. Analysis of Cell-Biomaterial Interaction through Cellular Bridge Formation in the Interface between HGMSCs and CaP Bioceramics. Sci. Rep. 2020, 10, 16493. [Google Scholar] [CrossRef]
- Zhang, Y.; Shu, T.; Wang, S.; Liu, Z.; Cheng, Y.; Li, A.; Pei, D. The Osteoinductivity of Calcium Phosphate-Based Biomaterials: A Tight Interaction With Bone Healing. Front. Bioeng. Biotechnol. 2022, 10, 911180. [Google Scholar] [CrossRef]
- Lai, W.; Chen, C.; Ren, X.; Lee, I.S.; Jiang, G.; Kong, X. Hydrothermal Fabrication of Porous Hollow Hydroxyapatite Microspheres for a Drug Delivery System. Mater. Sci. Eng. C 2016, 62, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Loca, D.; Locs, J.; Dubnika, A.; Zalite, V.; Berzina-Cimdina, L. Porous Hydroxyapatite for Drug Delivery; Elsevier Ltd.: Amsterdam, The Netherlands, 2015; ISBN 9781782420330. [Google Scholar]
- Niakan, A.; Ramesh, S.; Ganesan, P.; Tan, C.Y.; Purbolaksono, J.; Chandran, H.; Ramesh, S.; Teng, W.D. Sintering Behaviour of Natural Porous Hydroxyapatite Derived from Bovine Bone. Ceram. Int. 2015, 41, 3024–3029. [Google Scholar] [CrossRef]
- Sobczak, A.; Kowalski, Z. Metody Mokre Otrzymywania Hydroksyapatytu. Czas. Tech. 2008, 105, 125–131. [Google Scholar]
- Mohd Pu’ad, N.A.S.; Abdul Haq, R.H.; Mohd Noh, H.; Abdullah, H.Z.; Idris, M.I.; Lee, T.C. Synthesis Method of Hydroxyapatite: A Review. Mater. Today Proc. 2019, 29, 233–239. [Google Scholar] [CrossRef]
- Lugo, R.; Karthik, T.V.K.; Anaya, M.; Rosas, R.; Ceron, V.; Valderama, R.; Rodrguez, S. Wet Chemical Synthesis of Nanocrystalline Hydroxyapatite Flakes: Effect of PH and Sintering Temperature on Structural and Morphological Properties. R. Soc. Open Sci. 2018, 5, 1–14. [Google Scholar]
- Viswanath, B.; Ravishankar, N. Controlled Synthesis of Plate-Shaped Hydroxyapatite and Implications for the Morphology of the Apatite Phase in Bone. Biomaterials 2008, 29, 4855–4863. [Google Scholar] [CrossRef] [PubMed]
- Trzaskowska, M.; Vivcharenko, V.; Przekora, A. The Impact of Hydroxyapatite Sintering Temperature on Its Microstructural, Mechanical, and Biological Properties. Int. J. Mol. Sci. 2023, 24, 5083. [Google Scholar] [CrossRef]
- Wang, P.; Li, C.; Gong, H.; Jiang, X.; Wang, H.; Li, K. Effects of Synthesis Conditions on the Morphology of Hydroxyapatite Nanoparticles Produced by Wet Chemical Process. Powder Technol. 2010, 203, 315–321. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Ma, G.; Yang, D.; Nie, J. The Effect of the Prefrozen Process on Properties of a Chitosan/Hydroxyapatite/Poly(Methyl Methacrylate) Composite Prepared by Freeze Drying Method Used for Bone Tissue Engineering. RSC Adv. 2015, 5, 79679–79686. [Google Scholar] [CrossRef]
- Machowska, A.; Klara, J.; Ledwójcik, G.; Wójcik, K.; Dulińska-Litewka, J.; Karewicz, A. Clindamycin-Loaded Halloysite Nanotubes as the Antibacterial Component of Composite Hydrogel for Bone Repair. Polymers 2022, 14, 5151. [Google Scholar] [CrossRef]
- Manzano-Moreno, F.J.; Gónzalez-Acedo, A.; de Luna-Bertos, E.; García-Recio, E.; Ruiz, C.; Reyes-Botella, C. Effect of Amoxicillin and Clindamycin on the Gene Expression of Markers Involved in Osteoblast Physiology. J. Dent. Sci. 2023, 2. [Google Scholar] [CrossRef]
- Thadepalli, H.; Dhawan, V.K. Clindamycin A Review of Fifteen Years of Experience. Rev. Infect. Dis. 1982, 4, 1133–1153. [Google Scholar] [CrossRef]
- Gowri, M.; Latha, N.; Suganya, K.; Murugan, M.; Rajan, M. Calcium Alginate Nanoparticle Crosslinked Phosphorylated Polyallylamine to the Controlled Release of Clindamycin for Osteomyelitis Treatment. Drug Dev. Ind. Pharm. 2021, 47, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Smieja, M. Current Indications for the Use of Clindamycin: A Critical Review. Can. J. Infect. Dis. 1998, 9, 22–28. [Google Scholar] [CrossRef]
- Santamaría Arrieta, G.; Rodríguez Sánchez, F.; Rodriguez-Andrés, C.; Barbier, L.; Arteagoitia, I. The Effect of Preoperative Clindamycin in Reducing Early Oral Implant Failure: A Randomised Placebo-Controlled Clinical Trial. Clin. Oral Investig. 2023, 27, 1113–1122. [Google Scholar] [CrossRef]
- Arteagoitia, I.; Sánchez, F.R.; Figueras, A.; Arroyo-Lamas, N. Is Clindamycin Effective in Preventing Infectious Complications after Oral Surgery? Systematic Review and Meta-Analysis of Randomized Controlled Trials. Clin. Oral Investig. 2022, 26, 4467–4478. [Google Scholar] [CrossRef] [PubMed]
- Addy, L.D.; Martin, M.V. Clindamycin and Dentistry. Br. Dent. J. 2005, 199, 23–26. [Google Scholar] [CrossRef]
- Tancawan, A.L.; Pato, M.N.; Abidin, K.Z.; Asari, A.S.M.; Thong, T.X.; Kochhar, P.; Muganurmath, C.; Twynholm, M.; Barker, K. Amoxicillin/Clavulanic Acid for the Treatment of Odontogenic Infections: A Randomised Study Comparing Efficacy and Tolerability versus Clindamycin. Int. J. Dent. 2015, 2015, 472470. [Google Scholar] [CrossRef]
- Florkiewicz, W.; Słota, D.; Placek, A.; Pluta, K.; Tyliszczak, B.; Douglas, T.E.L.; Sobczak-Kupiec, A. Synthesis and Characterization of Polymer-Based Coatings Modified with Bioactive Ceramic and Bovine Serum Albumin. J. Funct. Biomater. 2021, 12, 21. [Google Scholar] [CrossRef]
- Słota, D.; Florkiewicz, W.; Piętak, K.; Szwed, A.; Włodarczyk, M.; Siwińska, M.; Rudnicka, K.; Sobczak-Kupiec, A. Preparation, Characterization, and Biocompatibility Assessment of Polymer-Ceramic Composites Loaded with Salvia Officinalis Extract. Materials 2021, 14, 6000. [Google Scholar] [CrossRef]
- Tomala, A.M.; Słota, D.; Florkiewicz, W.; Piętak, K.; Dylag, M.; Sobczak-Kupiec, A. Tribological Properties and Physiochemical Analysis of Polymer-Ceramic Composite Coatings for Bone Regeneration. Lubricants 2022, 10, 58. [Google Scholar] [CrossRef]
- Słota, D.; Głąb, M.; Tyliszczak, B.; Dogulas, T.E.L.; Rudnicka, K.; Miernik, K.; Urbaniak, M.M.; Rusek-wala, P.; Sobczak-kupiec, A. Composites Based on Hydroxyapatite and Whey Protein Isolate for Applications in Bone Regeneration. Materials 2021, 14, 2317. [Google Scholar] [CrossRef] [PubMed]
- Słota, D.; Piętak, K.; Florkiewicz, W.; Jampílek, J.; Tomala, A.; Urbaniak, M.M.; Tomaszewska, A.; Rudnicka, K.; Sobczak-Kupiec, A. Clindamycin-Loaded Nanosized Calcium Phosphates Powders as a Carrier of Active Substances. Nanomaterials 2023, 13, 1469. [Google Scholar] [CrossRef] [PubMed]
- Słota, D.; Florkiewicz, W.; Piętak, K.; Pluta, K.; Sadlik, J.; Miernik, K.; Sobczak-Kupiec, A. Preparation of PVP and Betaine Biomaterials Enriched with Hydroxyapatite and Its Evaluation as a Drug Carrier for Controlled Release of Clindamycin. Ceram. Int. 2022, 48, 35467–35473. [Google Scholar] [CrossRef]
- Słota, D.; Florkiewicz, W.; Sobczak-Kupiec, A. Ceramic-Polymer Coatings on Ti-6Al-4V Alloy Modified with L-Cysteine in Biomedical Applications. Mater. Today Commun. 2020, 25, 101301. [Google Scholar] [CrossRef]
- Reyes-Gasga, J.; Martínez-Piñeiro, E.L.; Rodríguez-Álvarez, G.; Tiznado-Orozco, G.E.; García-García, R.; Brès, E.F. XRD and FTIR Crystallinity Indices in Sound Human Tooth Enamel and Synthetic Hydroxyapatite. Mater. Sci. Eng. C 2013, 33, 4568–4574. [Google Scholar] [CrossRef]
- Zhou, W.Y.; Wang, M.; Cheung, W.L.; Guo, B.C.; Jia, D.M. Synthesis of Carbonated Hydroxyapatite Nanospheres through Nanoemulsion. J. Mater. Sci. Mater. Med. 2008, 19, 103–110. [Google Scholar] [CrossRef]
- Ślósarczyk, A.; Paszkiewicz, Z.; Paluszkiewicz, C. FTIR and XRD Evaluation of Carbonated Hydroxyapatite Powders Synthesized by Wet Methods. J. Mol. Struct. 2005, 744–747, 657–661. [Google Scholar] [CrossRef]
- Abifarin, J.K.; Obada, D.O.; Dauda, E.T.; Dodoo-Arhin, D. Experimental Data on the Characterization of Hydroxyapatite Synthesized from Biowastes. Data Br. 2019, 26, 104485. [Google Scholar] [CrossRef]
- Slimanou, H.; Baziz, A.; Bouzidi, N.; Quesada, D.E.; Tahakourt, A. Thermal, Physical, Mechanical and Microstructural Properties of Dredged Sediment-Based Ceramic Tiles as Substituent of Kaolin. Environ. Sci. Pollut. Res. 2022, 29, 26792–26809. [Google Scholar] [CrossRef]
- Farzan, M.; Roth, R.; Schoelkopf, J.; Huwyler, J.; Puchkov, M. The Processes behind Drug Loading and Release in Porous Drug Delivery Systems. Eur. J. Pharm. Biopharm. 2023, 189, 133–151. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Unit | d-HA | f-HA |
|---|---|---|---|
| drz | g/cm3 | 2.69 ± 0.08 | 2.44 ± 0.12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niziołek, K.; Słota, D.; Sadlik, J.; Łachut, E.; Florkiewicz, W.; Sobczak-Kupiec, A. Influence of Drying Technique on Physicochemical Properties of Synthetic Hydroxyapatite and Its Potential Use as a Drug Carrier. Materials 2023, 16, 6431. https://doi.org/10.3390/ma16196431
Niziołek K, Słota D, Sadlik J, Łachut E, Florkiewicz W, Sobczak-Kupiec A. Influence of Drying Technique on Physicochemical Properties of Synthetic Hydroxyapatite and Its Potential Use as a Drug Carrier. Materials. 2023; 16(19):6431. https://doi.org/10.3390/ma16196431
Chicago/Turabian StyleNiziołek, Karina, Dagmara Słota, Julia Sadlik, Emilia Łachut, Wioletta Florkiewicz, and Agnieszka Sobczak-Kupiec. 2023. "Influence of Drying Technique on Physicochemical Properties of Synthetic Hydroxyapatite and Its Potential Use as a Drug Carrier" Materials 16, no. 19: 6431. https://doi.org/10.3390/ma16196431