
Electronic, Thermal and Mechanical Properties of Carbon and Boron Nitride Holey Graphyne Monolayers


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussions
4. Concluding Remarks
- (a)
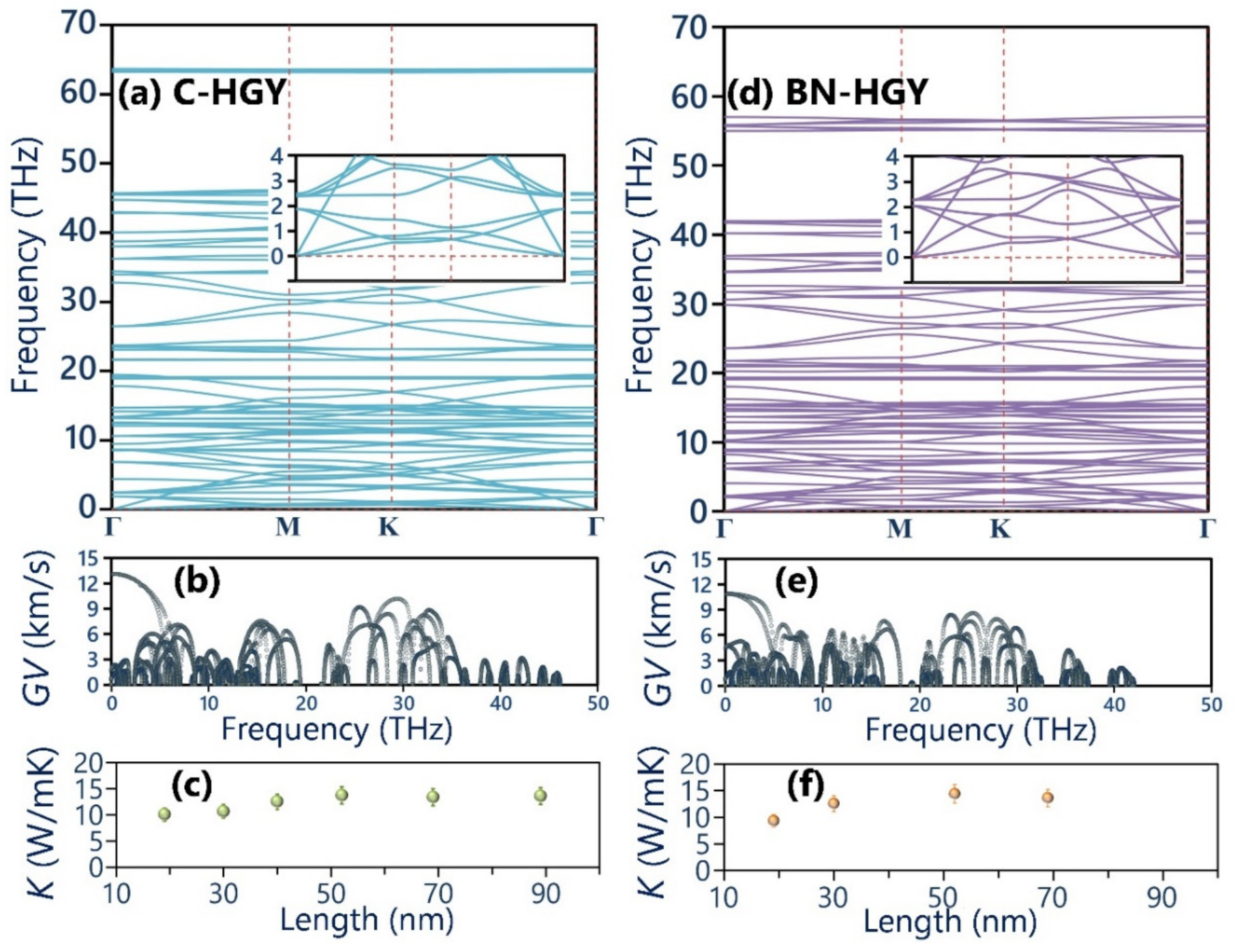
- Predicted phonon dispersion curves confirm the desired dynamical stability of the suspended C- and BN-HGY monolayers.
- (b)
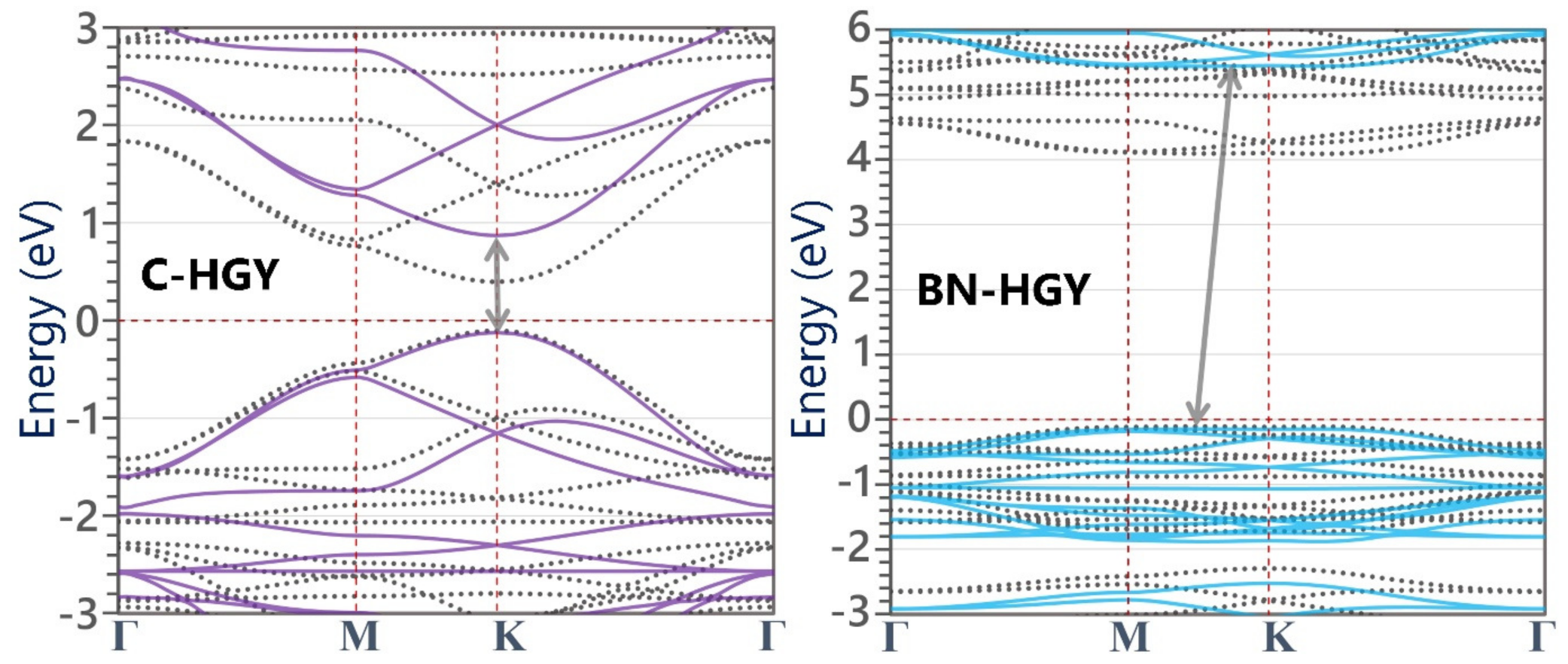
- The C-HGY monolayer is a direct gap semiconductor with 1.00 (0.50) eV gaps, according to the HSE06(PBE) functional.
- (c)
- The BN-HGY monolayer is predicted to be an insulator with low carrier mobilities due to almost flat dispersions of VBM and CBM.
- (d)
- The room temperature lattice thermal conductivity of the suspended C- and BN-HGY nanosheets is estimated to be very close, around 14.0 ± 1.5 W/mK.
- (e)
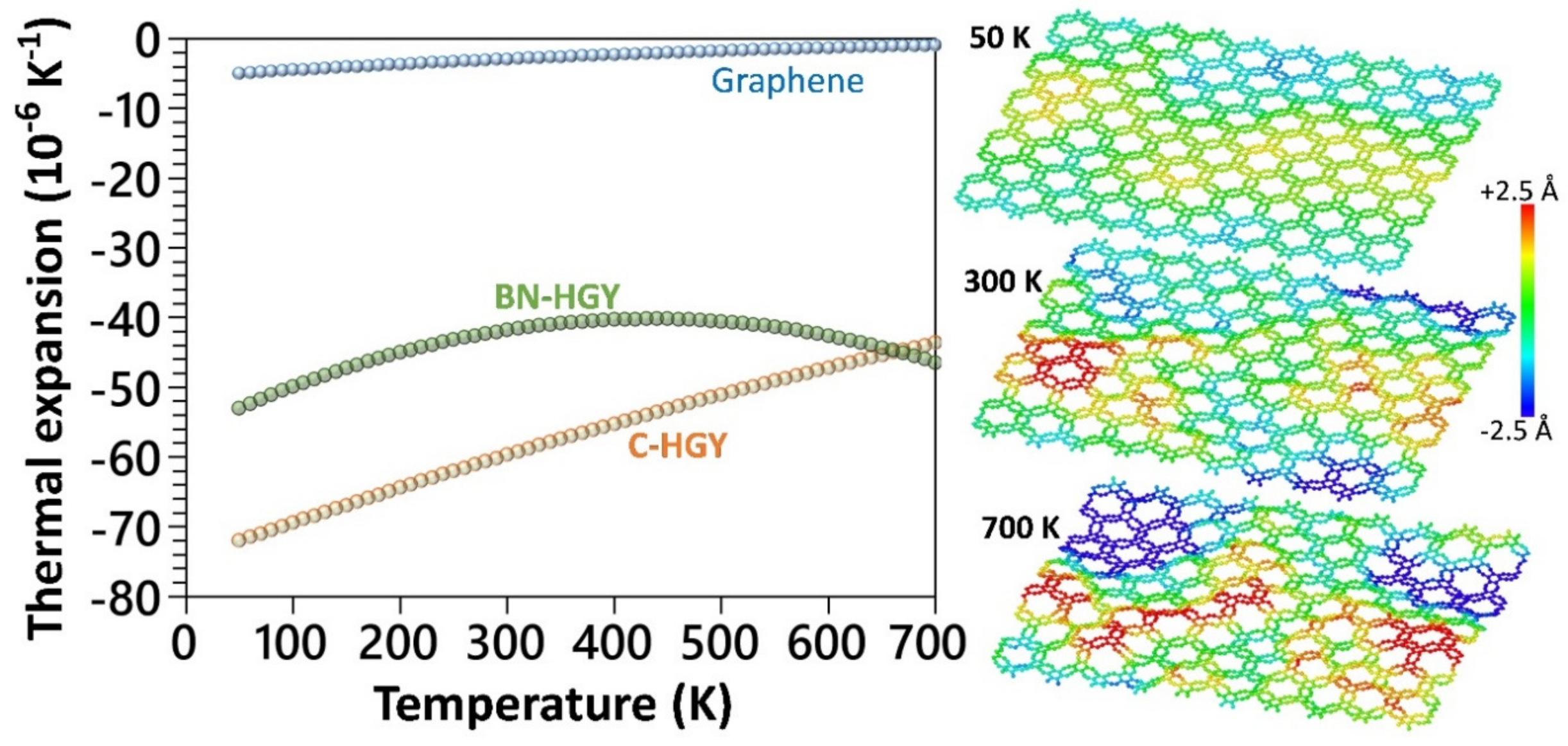
- The thermal expansion coefficient of the C- and BN-HGY nanomembranes at room temperature are predicted to be by more than one order of magnitude larger than that of the graphene.
- (f)
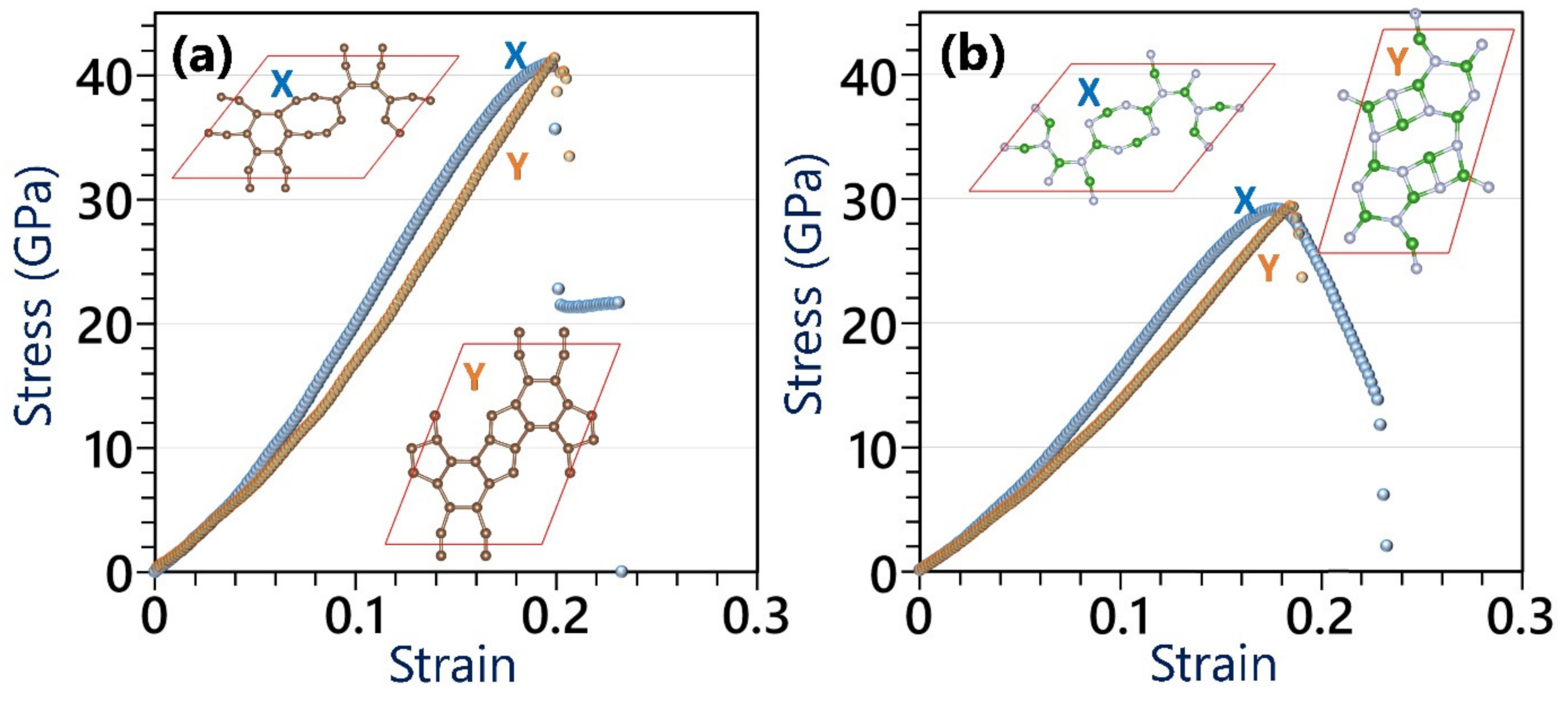
- The elastic modulus (ultimate tensile strength) values of the single-layer C- and BN-HGY are predicted to be 127(41) and 105(29) GPa, respectively.
- (g)
- The elastic modulus to ultimate tensile strength ratio of these nanoporous lattices at the ground state is predicted to be around 3, which is almost one third of that of other prominent 2D materials.
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Castro Neto, A.H.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K.; Guinea, F. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the Elastic Properties and Intrinsic Strength of Monolayer Graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Calizo, I.; Teweldebrhan, D.; Pokatilov, E.P.; Nika, D.L.; Balandin, A.A.; Bao, W.; Miao, F.; Lau, C.N. Extremely high thermal conductivity of graphene: Prospects for thermal management applications in nanoelectronic circuits. Appl. Phys. Lett. 2008, 92, 151911. [Google Scholar] [CrossRef]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior thermal conductivity of single-layer graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Banszerus, L.; Schmitz, M.; Engels, S.; Dauber, J.; Oellers, M.; Haupt, F.; Watanabe, K.; Taniguchi, T.; Beschoten, B.; Stampfer, C. Ultrahigh-mobility graphene devices from chemical vapor deposition on reusable copper. Sci. Adv. 2015, 1, e1500222. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Song, Z.; Li, T.; Li, X.; Ogbazghi, A.Y.; Feng, R.; Dai, Z.; Marchenkov, A.N.; Conrad, E.H.; First, P.N.; et al. Ultrathin Epitaxial Graphite: 2D Electron Gas Properties and a Route toward Graphene-based Nanoelectronics. J. Phys. Chem. B 2004, 108, 19912–19916. [Google Scholar] [CrossRef]
- Liu, M.; Yin, X.; Ulin-Avila, E.; Geng, B.; Zentgraf, T.; Ju, L.; Wang, F.; Zhang, X. A graphene-based broadband optical modulator. Nature 2011, 474, 64–67. [Google Scholar] [CrossRef]
- Withers, F.; Dubois, M.; Savchenko, A.K. Electron properties of fluorinated single-layer graphene transistors. Phys. Rev. B Condens. Matter Mater. Phys. 2010, 82, 73403. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, K. Recent progress on graphene-analogous 2D nanomaterials: Properties, modeling and applications. Prog. Mater. Sci. 2019, 100, 99–169. [Google Scholar] [CrossRef]
- Fan, Q.; Yan, L.; Tripp, M.W.; Krejčí, O.; Dimosthenous, S.; Kachel, S.R.; Chen, M.; Foster, A.S.; Koert, U.; Liljeroth, P.; et al. Biphenylene network: A nonbenzenoid carbon allotrope. Science 2021, 372, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Brunetto, G.; Autreto, P.A.S.; MacHado, L.D.; Santos, B.I.; Dos Santos, R.P.B.; Galvão, D.S. Nonzero gap two-dimensional carbon allotrope from porous graphene. J. Phys. Chem. C 2012, 116, 12810–12813. [Google Scholar] [CrossRef]
- Wang, X.-Q.; Li, H.-D.; Wang, J.-T. Prediction of a new two-dimensional metallic carbon allotrope. Phys. Chem. Chem. Phys. 2013, 15, 2024–2030. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, X.F.; Zhang, X.; Zhu, Q.; Dong, H.; Zhao, M.; Oganov, A.R. Phagraphene: A Low-Energy Graphene Allotrope Composed of 5-6-7 Carbon Rings with Distorted Dirac Cones. Nano Lett. 2015, 15, 6182–6186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, J.; Wang, Q.; Chen, X.; Kawazoe, Y.; Jena, P. Penta-graphene: A new carbon allotrope. Proc. Natl. Acad. Sci. USA 2015, 112, 2372–2377. [Google Scholar] [CrossRef]
- Baughman, R.H.; Eckhardt, H.; Kertesz, M. Structure-property predictions for new planar forms of carbon: Layered phases containing sp2 and sp atoms. J. Chem. Phys. 1987, 87, 6687. [Google Scholar] [CrossRef]
- Pan, L.D.; Zhang, L.Z.; Song, B.Q.; Du, S.X.; Gao, H.J. Graphyne- and graphdiyne-based nanoribbons: Density functional theory calculations of electronic structures. Appl. Phys. Lett. 2011, 98, 173102. [Google Scholar] [CrossRef]
- Li, G.; Li, Y.; Liu, H.; Guo, Y.; Li, Y.; Zhu, D. Architecture of graphdiyne nanoscale films. Chem. Commun. 2010, 46, 3256–3258. [Google Scholar] [CrossRef]
- Matsuoka, R.; Sakamoto, R.; Hoshiko, K.; Sasaki, S.; Masunaga, H.; Nagashio, K.; Nishihara, H. Crystalline Graphdiyne Nanosheets Produced at a Gas/Liquid or Liquid/Liquid Interface. J. Am. Chem. Soc. 2017, 139, 3145–3152. [Google Scholar] [CrossRef]
- Kan, X.; Ban, Y.; Wu, C.; Pan, Q.; Liu, H.; Song, J.; Zuo, Z.; Li, Z.; Zhao, Y. Interfacial Synthesis of Conjugated Two-Dimensional N-Graphdiyne. ACS Appl. Mater. Interfaces 2018, 10, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Li, X.; Tu, Z.; Zhao, F.; He, J.; Guan, Z.; Huang, C.; Yi, Y.; Li, Y. Synthesis, Electronic Structure of Boron-Graphdiyne with an sp-Hybridized Carbon Skeleton and Its Application in Sodium Storage. Angew. Chemie 2018, 130, 4169. [Google Scholar] [CrossRef]
- Matsuoka, R.; Toyoda, R.; Shiotsuki, R.; Fukui, N.; Wada, K.; Maeda, H.; Sakamoto, R.; Sasaki, S.; Masunaga, H.; Nagashio, K.; et al. Expansion of the Graphdiyne Family: A Triphenylene-Cored Analogue. ACS Appl. Mater. Interfaces 2018, 11, 2730–2733. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wu, C.; Pan, Q.; Jin, Y.; Lyu, R.; Martinez, V.; Huang, S.; Wu, J.; Wayment, L.J.; Clark, N.A.; et al. Synthesis of γ-graphyne using dynamic covalent chemistry. Nat. Synth. 2022, 1, 449–454. [Google Scholar] [CrossRef]
- Liu, X.; Cho, S.M.; Lin, S.; Chen, Z.; Choi, W.; Kim, Y.-M.; Yun, E.; Baek, E.H.; Ryu, D.H.; Lee, H. Constructing two-dimensional holey graphyne with unusual annulative π-extension. Matter 2022, 5, 2306–2318. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Monkhorst, H.; Pack, J. Special points for Brillouin zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Shapeev, A.V. Moment tensor potentials: A class of systematically improvable interatomic potentials. Multiscale Model. Simul. 2016, 14, 1153–1173. [Google Scholar] [CrossRef]
- Novikov, I.S.; Gubaev, K.; Podryabinkin, E.V.; Shapeev, A.V. The MLIP package: Moment Tensor Potentials with MPI and Active Learning. Mach. Learn. Sci. Technol. 2021, 2, 025002. [Google Scholar] [CrossRef]
- Mortazavi, B.; Silani, M.; Podryabinkin, E.V.; Rabczuk, T.; Zhuang, X.; Shapeev, A. V First-Principles Multiscale Modeling of Mechanical Properties in Graphene/Borophene Heterostructures Empowered by Machine-Learning Interatomic Potentials. Adv. Mater. 2021, 33, 2102807. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, B.; Shojaei, F.; Shapeev, A.V.; Zhuang, X. A combined first-principles and machine-learning investigation on the stability, electronic, optical, and mechanical properties of novel C6N7-based nanoporous carbon nitrides. Carbon N. Y. 2022, 194, 230–239. [Google Scholar] [CrossRef]
- Mortazavi, B.; Podryabinkin, E.V.; Roche, S.; Rabczuk, T.; Zhuang, X.; Shapeev, A. V Machine-learning interatomic potentials enable first-principles multiscale modeling of lattice thermal conductivity in graphene/borophene heterostructures. Mater. Horizons 2020, 7, 2359–2367. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Mortazavi, B.; Novikov, I.S.; Podryabinkin, E.V.; Roche, S.; Rabczuk, T.; Shapeev, A.V.; Zhuang, X. Exploring phononic properties of two-dimensional materials using machine learning interatomic potentials. Appl. Mater. Today 2020, 20, 100685. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Sajjad, M.; Nair, S.S.; Samad, Y.A.; Singh, N. Colossal figure of merit and compelling HER catalytic activity of holey graphyne. Sci. Rep. 2023, 13, 9123. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of Chemical-Bonds Based on Topological Analysis of Electron Localization Functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Tromer, R.M.; Felix, I.M.; Pereira, L.; da Luz, M.E.G.G.E.; Ribeiro, L.A.; Galvao, D.S. Lattice Thermal Conductivity of 2D Nanomaterials: A Simple Semi-Empirical Approach. Phys. Chem. Chem. Phys. 2023. [Google Scholar] [CrossRef]
- Mortazavi, B.; Rajabpour, A.; Zhuang, X.; Rabczuk, T.; Shapeev, A. V Exploring thermal expansion of carbon-based nanosheets by machine-learning interatomic potentials. Carbon N. Y. 2022, 186, 501–508. [Google Scholar] [CrossRef]
- Ghasemi, H.; Rajabpour, A. A novel approach to calculate thermal expansion of graphene: Molecular dynamics study. Eur. Phys. J. Plus 2017, 132, 221. [Google Scholar] [CrossRef]
- Griffith, A.A. The Phenomena of Rupture and Flow in Solids. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 1921, 221, 163–198. [Google Scholar] [CrossRef]
- Bertolazzi, S.; Brivio, J.; Kis, A. Stretching and breaking of ultrathin MoS 2. ACS Nano 2011, 5, 9703–9709. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, B.; Rahaman, O.; Makaremi, M.; Dianat, A.; Cuniberti, G.; Rabczuk, T. First-principles investigation of mechanical properties of silicene, germanene and stanene. Phys. E Low-Dimens. Syst. Nanostructures 2017, 87, 228–232. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mortazavi, B. Electronic, Thermal and Mechanical Properties of Carbon and Boron Nitride Holey Graphyne Monolayers. Materials 2023, 16, 6642. https://doi.org/10.3390/ma16206642
Mortazavi B. Electronic, Thermal and Mechanical Properties of Carbon and Boron Nitride Holey Graphyne Monolayers. Materials. 2023; 16(20):6642. https://doi.org/10.3390/ma16206642
Chicago/Turabian StyleMortazavi, Bohayra. 2023. "Electronic, Thermal and Mechanical Properties of Carbon and Boron Nitride Holey Graphyne Monolayers" Materials 16, no. 20: 6642. https://doi.org/10.3390/ma16206642
APA StyleMortazavi, B. (2023). Electronic, Thermal and Mechanical Properties of Carbon and Boron Nitride Holey Graphyne Monolayers. Materials, 16(20), 6642. https://doi.org/10.3390/ma16206642