
Study on Synergistic Effect of Xanthan Gum and Sodium Methylsiliconate on Mechanical Strength and Water Stability of Phosphogypsum Road-Based Materials



Abstract
:1. Introduction
2. Materials and Experimental Procedure
2.1. Materials
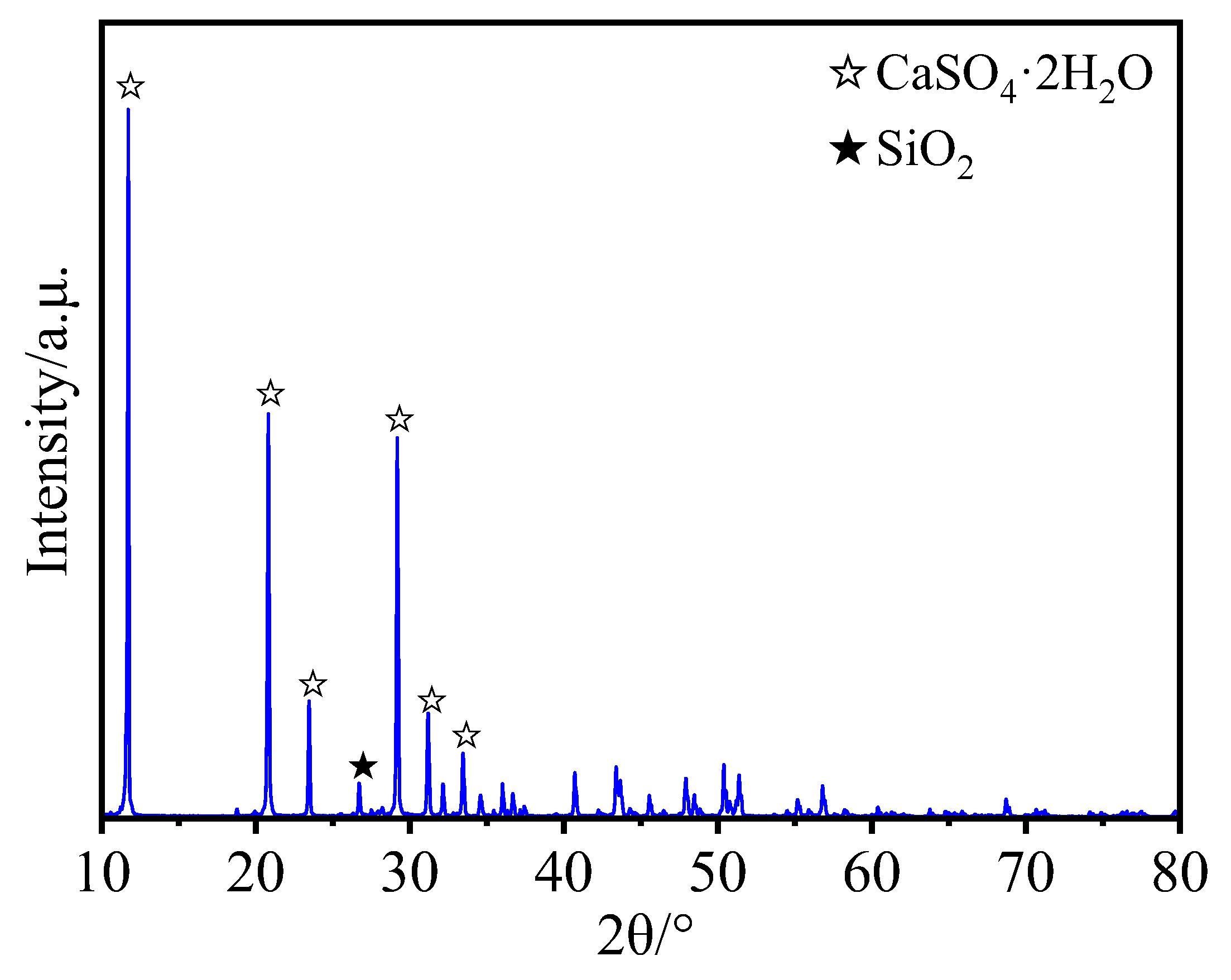
2.1.1. PG
2.1.2. Curing Agents
2.2. Experimental Procedure
2.2.1. Preparation of Stabilized PG
2.2.2. Unconfined Compressive Strength
2.2.3. Water Stability
2.2.4. Leaching of Hazardous Substance
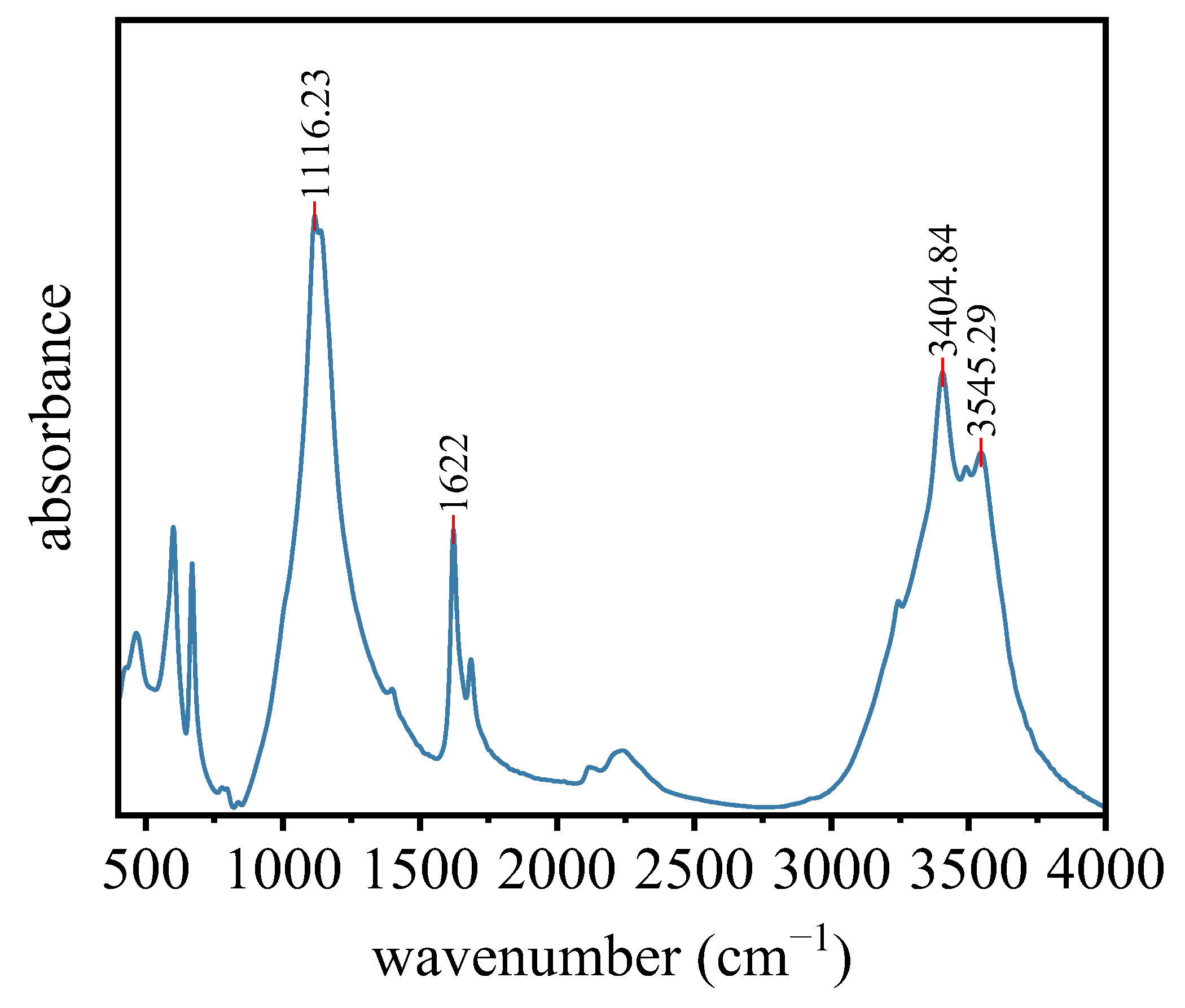
2.2.5. Other Characterization Methods
3. Results and Discussion
3.1. Unconfined Compressive Strength
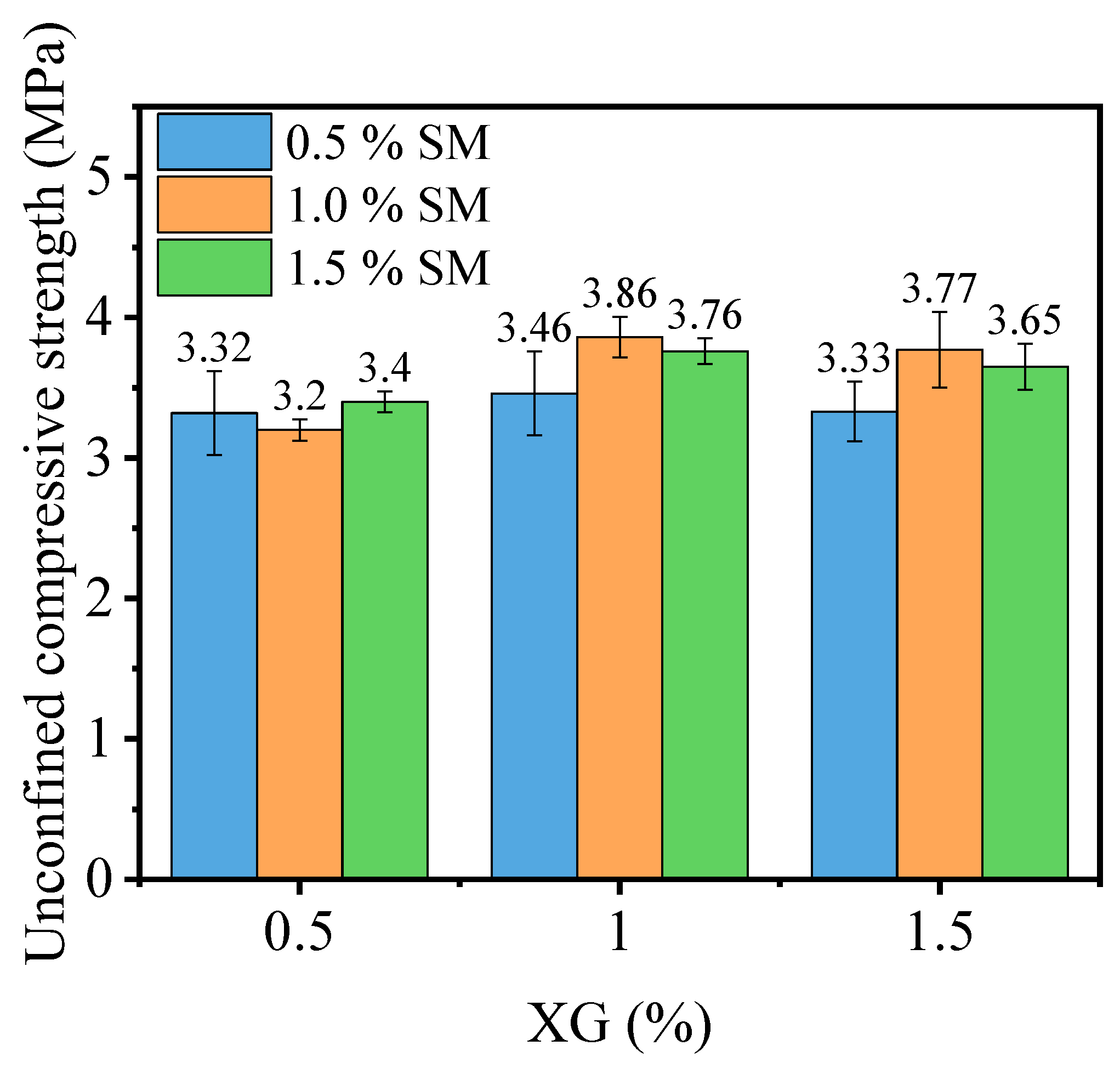
3.1.1. Effect of Curing Agent Dosage
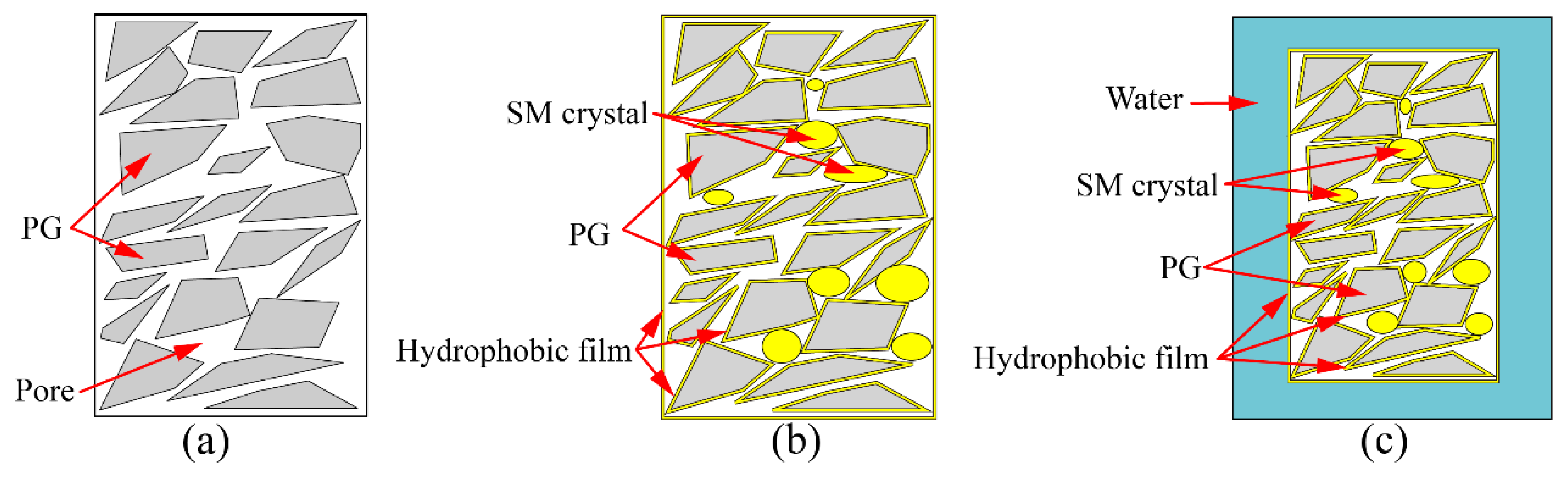
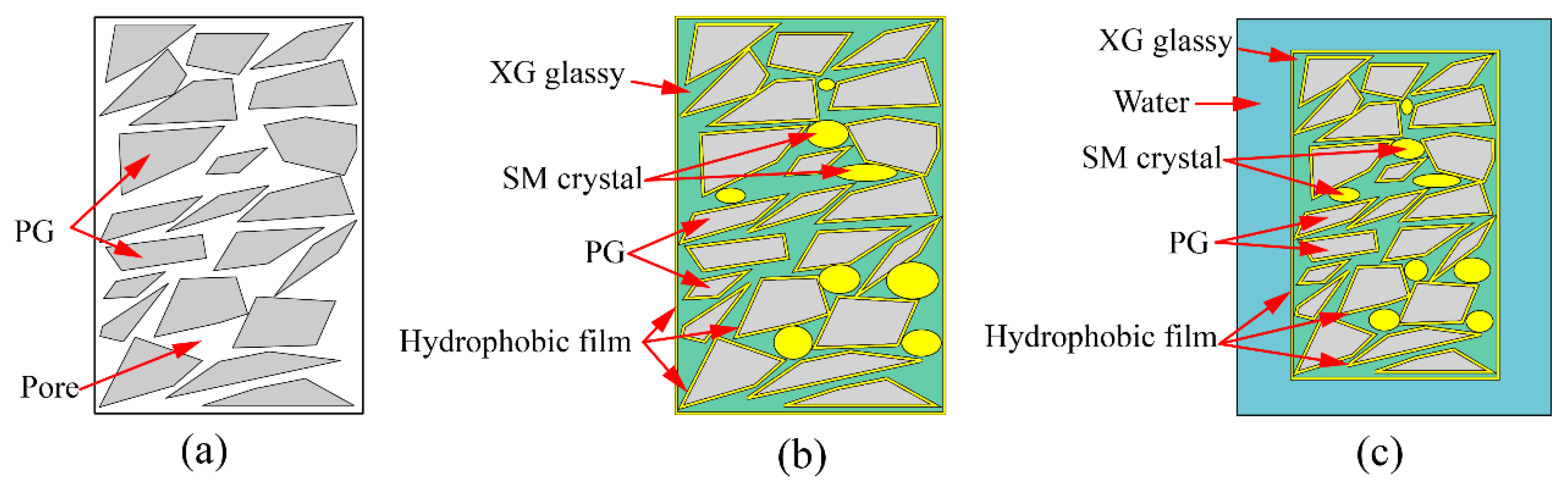
3.1.2. Mechanism of Unconfined Compressive Strength Increasing
3.1.3. Effect of Temperature and Curing Time
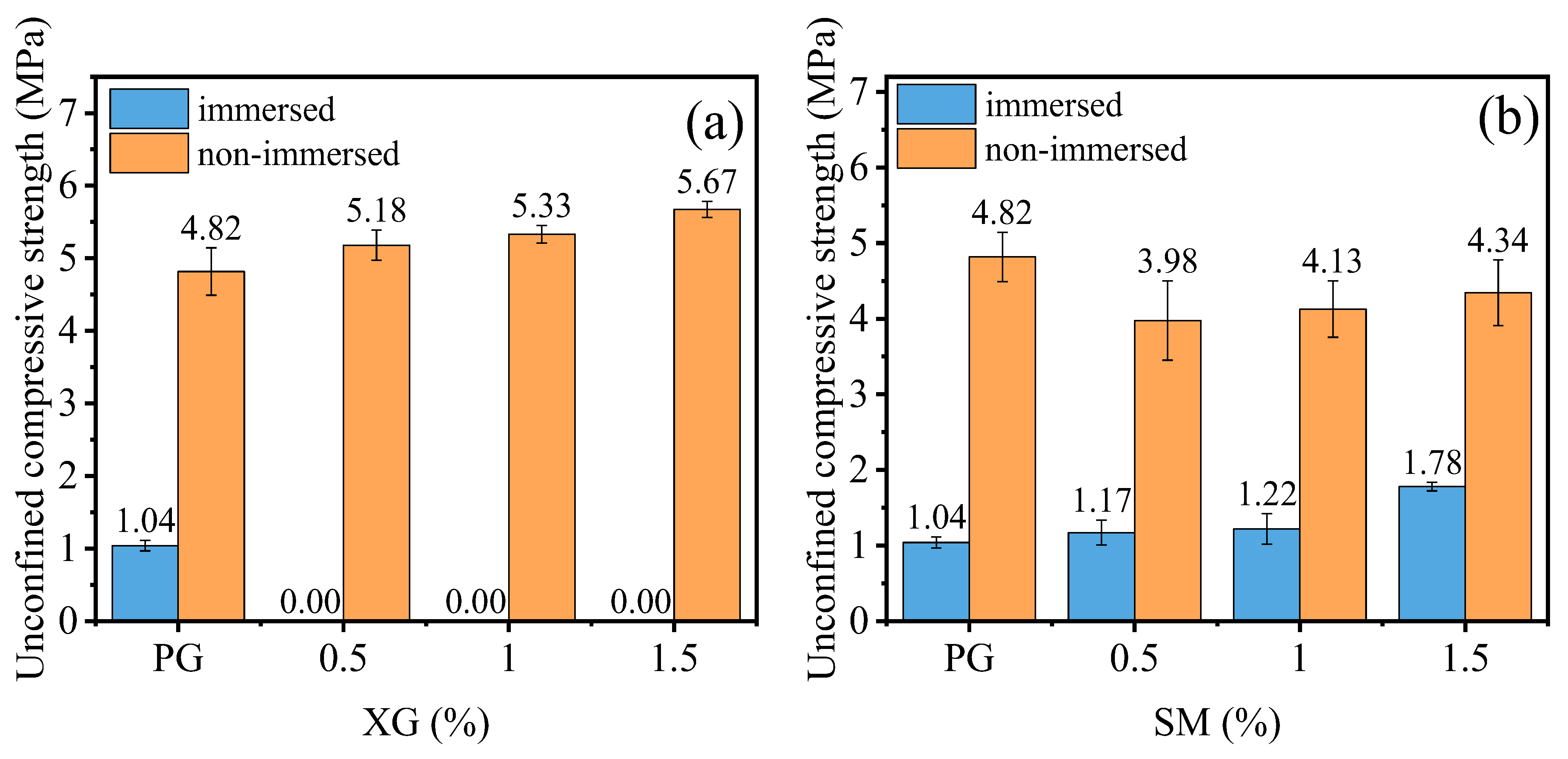
3.2. Water Stability
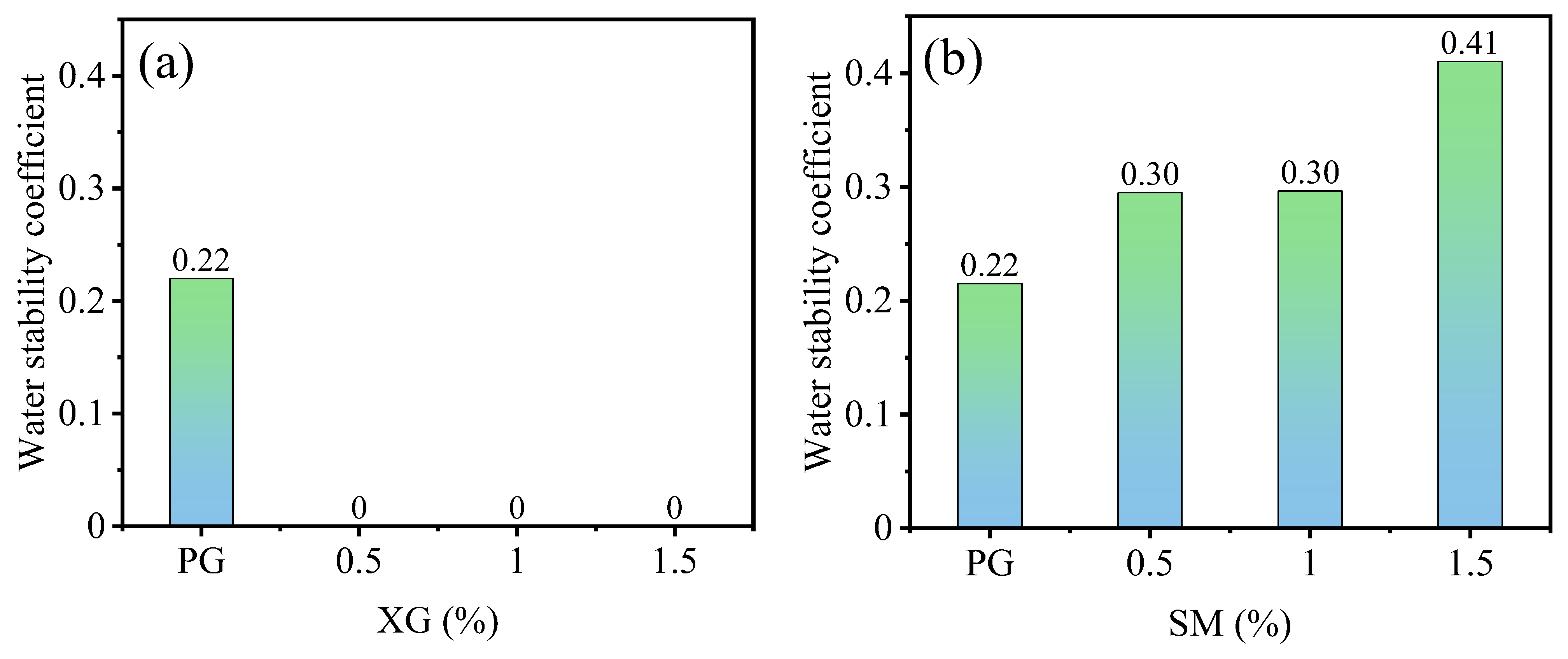
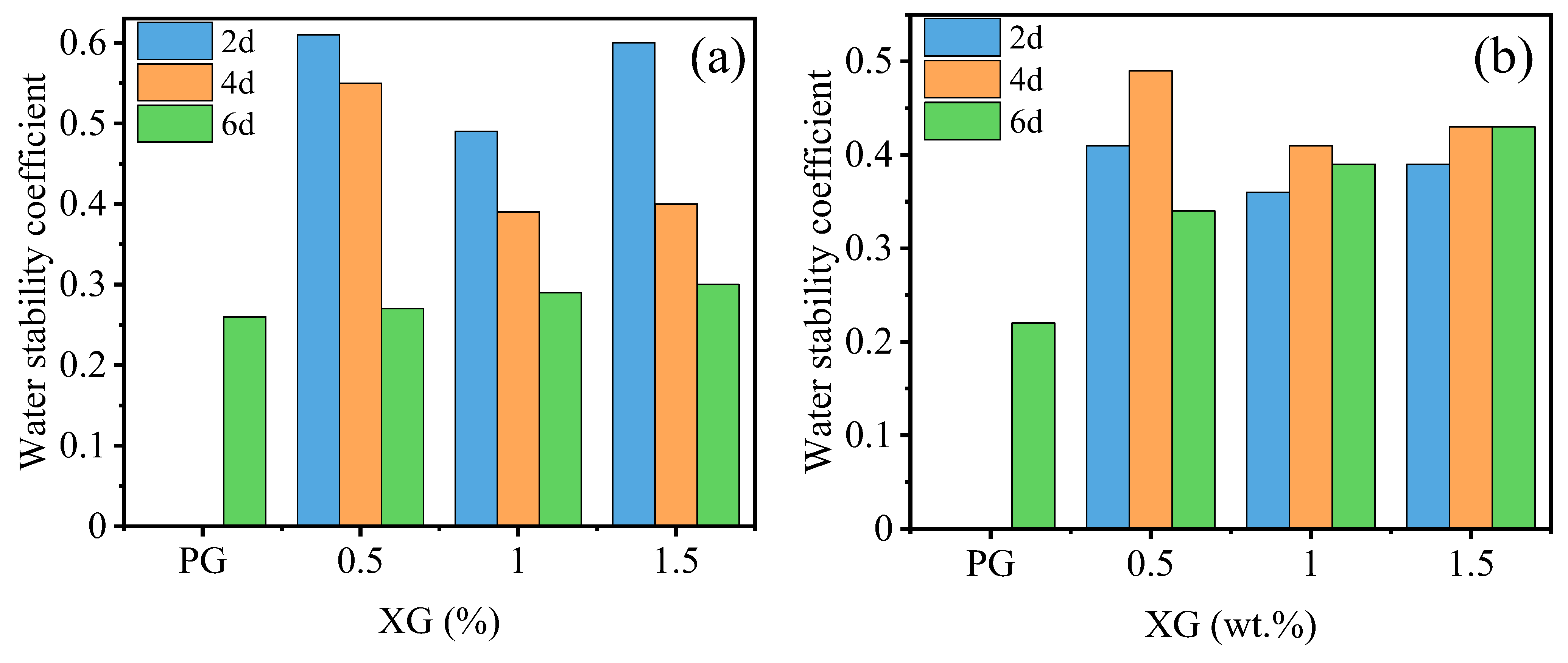
3.2.1. Effect of Curing Agent Dosage
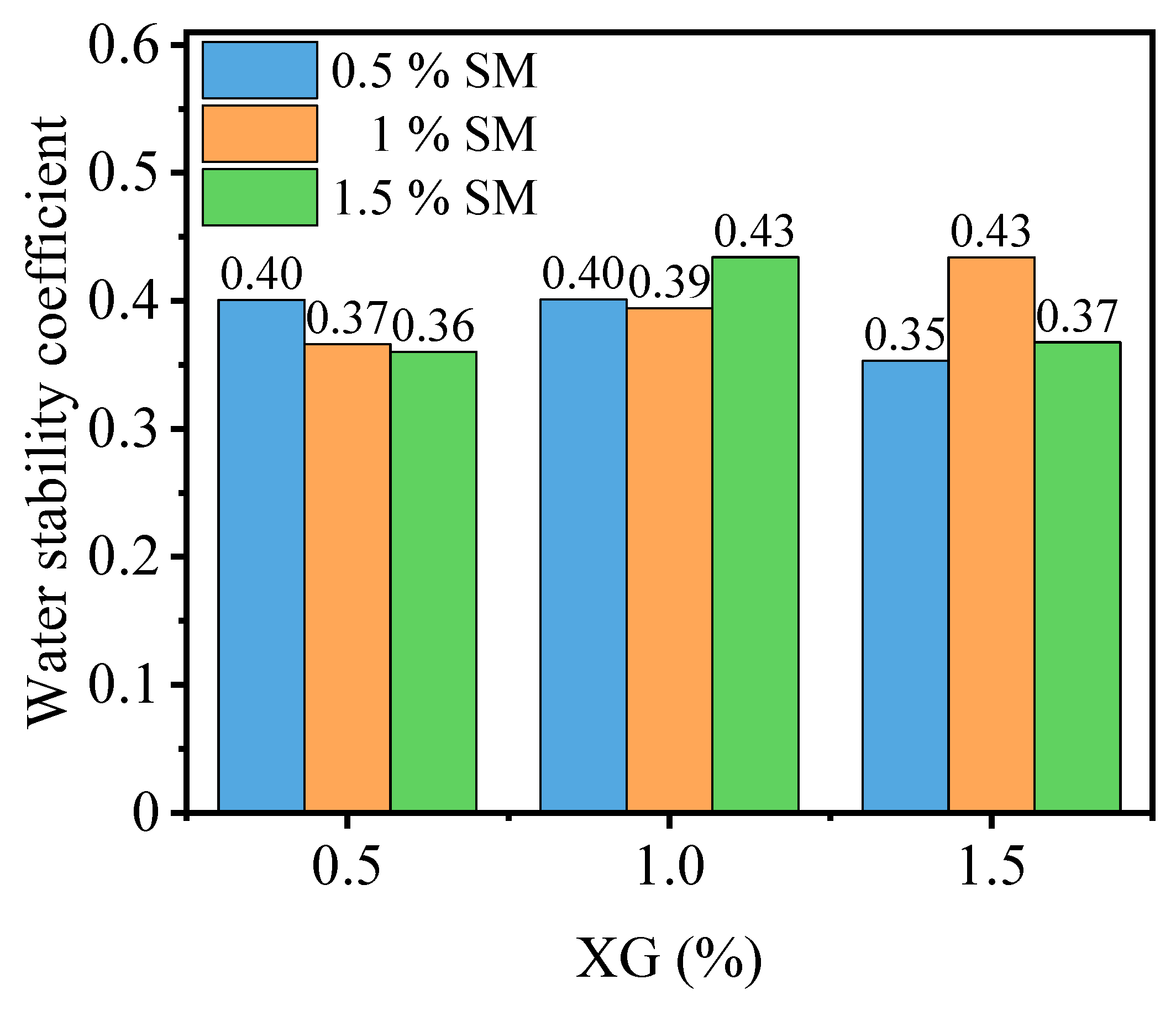
3.2.2. Effect of Temperature and Curing Time
3.3. Leaching of Hazardous Substance
4. Conclusions
- (1)
- Incorporating XG and SM together in PG yields positive effects on its unconfined compressive strength, water stability coefficient, and the reduction of hazardous substance leaching. Notably, the addition of SM improves strength and water stability, whereas the addition of XG decreases the strength and weakens water stability. The most favorable outcomes can be obtained when the concentration is 1.0% for XG and SM individually. The presence of a glassy material formed by XG fills the voids in PG and bonds the particles together, while the hydrophobic film formed by SM protects the PG and XG from water erosion, leading to improved water stability and strength of stabilized PG.
- (2)
- The strength of stabilized PG increases with curing time curing temperature, while the water stability coefficient exhibited varying trends depending on the curing temperature. The curing temperature and time significantly influence the rate of XG gel transforming into a glassy state, with faster transformation being more favorable.
- (3)
- The dual incorporation of XG and SM also remarkably reduces the leaching of hazardous substances from PG, particularly at both concentrations of 1.0%. This reduction is achieved through physical adsorption, chemical binding, and physical blocking of the release of hazardous substances by XG and SM.
- (4)
- The results verify the feasibility of XG and SM stabilized phosphogypsum roadbed materials, which are more environmentally friendly than traditional inorganic cementing materials, but there are also some defects. Future research can focus on improving the curing process, evaluating long-term performance, and assessing environmental impacts. Addressing these aspects will enable the utilization of XG and SM as curing agents for PG-based materials to offer environmentally friendly and economically feasible solutions for sustainable infrastructure development.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rashad, A.M. Phosphogypsum as a construction material. J. Clean. Prod. 2017, 166, 732–743. [Google Scholar] [CrossRef]
- Rashad, A.M. Potential use of phosphogypsum in alkali-activated fly ash under the effects of elevated temperatures and thermal shock cycles. J. Clean. Prod. 2015, 87, 717–725. [Google Scholar] [CrossRef]
- Chen, S.; Chen, J.; He, X.; Su, Y.; Jin, Z.; Fan, J.; Qi, H.; Wang, B. Comparative analysis of colloid-mechanical microenvironments on the efficient purification of phosphogypsum. Constr. Build. Mater. 2023, 392, 132037. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, F.; Ma, L.; Ning, P.; Yang, J.; Wei, Y. An efficient methodology to use hydrolysate of phosphogypsum decomposition products for CO2 mineral sequestration and calcium carbonate production. J. Clean. Prod. 2020, 259, 120826. [Google Scholar] [CrossRef]
- Tsioka, M.; Voudrias, E.A. Comparison of alternative management methods for phosphogypsum waste using life cycle analysis. J. Clean. Prod. 2020, 266, 121386. [Google Scholar] [CrossRef]
- Degirmenci, N.; Okucu, A.; Turabi, A. Application of phosphogypsum in soil stabilization. Build. Environ. 2007, 42, 3393–3398. [Google Scholar] [CrossRef]
- Hentati, O.; Abrantes, N.; Caetano, A.L.; Bouguerra, S.; Gonçalves, F.; Römbke, J.; Pereira, R. Phosphogypsum as a soil fertilizer: Ecotoxicity of amended soil and elutriates to bacteria, invertebrates, algae and plants. J. Hazard. Mater. 2015, 294, 80–89. [Google Scholar] [CrossRef]
- Michalovicz, L.; Tormena, C.A.; Müller, M.M.L.; Dick, W.A.; Cervi, E.C. Residual effects of phosphogypsum rates and machinery traffic on soil attributes and common-bean (Phaseolus vulgaris) yield in a no-tillage system. Soil Tillage Res. 2021, 213, 105152. [Google Scholar] [CrossRef]
- Huang, L.; Liu, Y.; Ferreira, J.F.S.; Wang, M.; Na, J.; Huang, J.; Liang, Z. Long-term combined effects of tillage and rice cultivation with phosphogypsum or farmyard manure on the concentration of salts, minerals, and heavy metals of saline-sodic paddy fields in Northeast China. Soil Tillage Res. 2022, 215, 105222. [Google Scholar] [CrossRef]
- Costa, R.F.; Firmano, R.F.; Colzato, M.; Crusciol, C.A.C.; Alleoni, L.R.F. Sulfur speciation in a tropical soil amended with lime and phosphogypsum under long-term no-tillage system. Geoderma 2022, 406, 115461. [Google Scholar] [CrossRef]
- Calderón-Morales, B.R.S.; García-Martínez, A.; Pineda, P.; García-Tenório, R. Valorization of phosphogypsum in cement-based materials: Limits and potential in eco-efficient construction. J. Build. Eng. 2021, 44, 102506. [Google Scholar] [CrossRef]
- Chen, M.; Liu, P.; Kong, D.; Wang, Y.; Wang, J.; Huang, Y.; Yu, K.; Wu, N. Influencing factors of mechanical and thermal conductivity of foamed phosphogypsum-based composite cementitious materials. Constr. Build. Mater. 2022, 346, 128462. [Google Scholar] [CrossRef]
- Sun, T.; Li, W.; Xu, F.; Yu, Z.; Wang, Z.; Ouyang, G.; Xu, D. A new eco-friendly concrete made of high content phosphogypsum based aggregates and binder: Mechanical properties and environmental benefits. J. Clean. Prod. 2023, 400, 136555. [Google Scholar] [CrossRef]
- Altun, I.A.; Sert, Y. Utilization of weathered phosphogypsum as set retarder in Portland cement. Cem. Concr. Res. 2004, 34, 677–680. [Google Scholar] [CrossRef]
- Potgieter, J.H.; Potgieter, S.S.; McCrindle, R.I.; Strydom, C.A. An investigation into the effect of various chemical and physical treatments of a South African phosphogypsum to render it suitable as a set retarder for cement. Cem. Concr. Res. 2003, 33, 1223–1227. [Google Scholar] [CrossRef]
- Costa, R.P.; de Medeiros, M.H.G.; Martinez, E.D.R.; Quarcioni, V.A.; Suzuki, S.; Kirchheim, A.P. Effect of soluble phosphate, fluoride, and pH in Brazilian phosphogypsum used as setting retarder on Portland cement hydration. Case Stud. Constr. Mater. 2022, 17, e1413. [Google Scholar] [CrossRef]
- Li, B.; Li, L.; Chen, X.; Ma, Y.; Zhou, M. Modification of phosphogypsum using circulating fluidized bed fly ash and carbide slag for use as cement retarder. Constr. Build. Mater. 2022, 338, 127630. [Google Scholar] [CrossRef]
- Ölmez, H.; Erdem, E. The effects of phosphogypsum on the setting and mechanical properties of Portland cement and trass cement. Cem. Concr. Res. 1989, 19, 377–384. [Google Scholar] [CrossRef]
- Taylor, D.C. Policy incentives to minimize generation of municipal solid waste. Waste Manag. Res. 2000, 18, 406–419. [Google Scholar] [CrossRef]
- Abdelbasir, S.M. Recycling, Management, and Valorization of Industrial Solid Wastes. In Waste Recycling Technologies for Nanomaterials Manufacturing; Makhlouf, A.S.H., Ali, G.A.M., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 25–63. [Google Scholar]
- Diouri, C.; Echehbani, I.; Lahlou, K.; Omari, K.E.; Alaoui, A. Valorization of Moroccan phosphogypsum in road engineering: Parametric study. Mater. Today Proc. 2022, 58, 1054–1058. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, D.; You, L.; Luo, H.; Xu, W. Recycling phosphogypsum in subbase of pavement: Treatment, testing, and application. Constr. Build. Mater. 2022, 342, 127948. [Google Scholar] [CrossRef]
- Amrani, M.; Taha, Y.; Kchikach, A.; Benzaazoua, M.; Hakkou, R. Phosphogypsum recycling: New horizons for a more sustainable road material application. J. Build. Eng. 2020, 30, 101267. [Google Scholar] [CrossRef]
- Men, J.; Li, Y.; Cheng, P.; Zhang, Z. Recycling phosphogypsum in road construction materials and associated environmental considerations: A review. Heliyon 2022, 8, e11518. [Google Scholar] [CrossRef]
- Qiao, D.; Qian, J.S.; Wang, Q.Z.; Dang, Y.D.; Zhang, H.; Zeng, D.Q. Utilization of sulfate-rich solid wastes in rural road construction in the Three Gorges Reservoir. Resour. Conserv. Recy. 2010, 54, 1368–1376. [Google Scholar] [CrossRef]
- Tayibi, H.; Choura, M.; López, F.A.; Alguacil, F.J.; López-Delgado, A. Environmental impact and management of phosphogypsum. J. Environ. Manag. 2009, 90, 2377–2386. [Google Scholar] [CrossRef]
- Reijnders, L. Cleaner phosphogypsum, coal combustion ashes and waste incineration ashes for application in building materials: A review. Build. Environ. 2007, 42, 1036–1042. [Google Scholar] [CrossRef]
- Prasad, M.N.V. Chapter 14–Resource Potential of Natural and Synthetic Gypsum Waste. In Environmental Materials and Waste; Prasad, M.N.V., Shih, K., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 307–337. [Google Scholar]
- Chernysh, Y.; Yakhnenko, O.; Chubur, V.; Roubík, H. Phosphogypsum Recycling: A Review of Environmental Issues, Current Trends, and Prospects. Appl. Sci. 2021, 11, 1575. [Google Scholar] [CrossRef]
- Qin, X.; Cao, Y.; Guan, H.; Hu, Q.; Liu, Z.; Xu, J.; Hu, B.; Zhang, Z.; Luo, R. Resource utilization and development of phosphogypsum-based materials in civil engineering. J. Clean. Prod. 2023, 387, 135858. [Google Scholar] [CrossRef]
- Moussa, D.; Crispel, J.J.; Legrand, C.; Thenoz, B. Laboratory study of the structure and compactibility of Tunisian phosphogypsum (SFAX) for use in embankment construction. Resour. Conserv. 1984, 11, 95–116. [Google Scholar] [CrossRef]
- Meskini, S.; Samdi, A.; Ejjaouani, H.; Remmal, T. Valorization of phosphogypsum as a road material: Stabilizing effect of fly ash and lime additives on strength and durability. J. Clean. Prod. 2021, 323, 129161. [Google Scholar] [CrossRef]
- Ngo, H.T.T.; Dang, V.Q.; Ho, L.S.; Doan, T.X. Utilization phosphogypsum as a construction material for road base: A case study in Vietnam. Innov. Infrastruct. Solut. 2021, 7, 88. [Google Scholar] [CrossRef]
- Shen, W.G.; Zhou, M.K.; Ma, W.; Hu, J.Q.; Cai, Z. Investigation on the application of steel slag-fly ash-phosphogypsum solidified material as road base material. J. Hazard. Mater. 2009, 164, 99–104. [Google Scholar] [CrossRef]
- Soldo, A.; Mileti, M.; Auad, M.L. Biopolymers as a sustainable solution for the enhancement of soil mechanical properties. Sci. Rep. 2020, 10, 267. [Google Scholar] [CrossRef]
- Huang, J.; Kogbara, R.B.; Hariharan, N.; Masad, E.A.; Little, D.N. A state-of-the-art review of polymers used in soil stabilization. Constr. Build. Mater. 2021, 305, 124685. [Google Scholar] [CrossRef]
- Ivanov, V.; Chu, J. Applications of microorganisms to geotechnical engineering for bioclogging and biocementation of soil in situ. Rev. Environ. Sci. Bio/Technol. 2008, 7, 139–153. [Google Scholar] [CrossRef]
- Swain, K.; Mahamaya, M.; Alam, S.; Das, S.K. Stabilization of Dispersive Soil Using Biopolymer; Springer International Publishing: Cham, Switzerland, 2018; pp. 132–147. [Google Scholar]
- Chen, C.; Wu, L.; Perdjon, M.; Huang, X.; Peng, Y. The drying effect on xanthan gum biopolymer treated sandy soil shear strength. Constr. Build. Mater. 2019, 197, 271–279. [Google Scholar] [CrossRef]
- Jang, H.Y.; Zhang, K.; Chon, B.H.; Choi, H.J. Enhanced oil recovery performance and viscosity characteristics of polysaccharide xanthan gum solution. J. Ind. Eng. Chem. 2015, 21, 741–745. [Google Scholar] [CrossRef]
- Vydehi, K.V.; Moghal, A.A.B. Effect of Biopolymeric Stabilization on the Strength and Compressibility Characteristics of Cohesive Soil. J. Mater. Civ. Eng. 2022, 34, 4021428. [Google Scholar] [CrossRef]
- Chang, I.; Im, J.; Prasidhi, A.K.; Cho, G. Effects of Xanthan gum biopolymer on soil strengthening. Constr. Build. Mater. 2015, 74, 65–72. [Google Scholar] [CrossRef]
- Nugent, R.A.; Zhang, G.; Gambrell, R.P. Effect of Exopolymers on the Liquid Limit of Clays and Its Engineering Implications. Transport. Res. Rec. 2009, 2101, 34–43. [Google Scholar] [CrossRef]
- Chang, I.; Prasidhi, A.K.; Im, J.; Cho, G. Soil strengthening using thermo-gelation biopolymers. Constr. Build. Mater. 2015, 77, 430–438. [Google Scholar] [CrossRef]
- American Public Health Association. Standard Methods for the Examination of Water and Wastewater; American Public Health Association: San Francisco, CA, USA, 1912. [Google Scholar]
- Yakimets, I.; Paes, S.S.; Wellner, N.; Smith, A.C.; Wilson, R.H.; Mitchell, J.R. Effect of Water Content on the Structural Reorganization and Elastic Properties of Biopolymer Films: A Comparative Study. Biomacromolecules 2007, 8, 1710–1722. [Google Scholar] [CrossRef]
- Huang, G.; Lin, H.; Li, J.; Liu, J. Inducing hydrophobicity in saline soils: A comparison of hydrophobic agents and mechanisms. Powder Technol. 2023, 424, 118475. [Google Scholar] [CrossRef]
- Ma, Q.; Liu, Q.; Cheng, K.; Liu, S. Study on the Durability of Hydraulic Lime Soil Mixed with Sodium Methyl Silicate. Coatings 2022, 12, 903. [Google Scholar] [CrossRef]
- Min, J.; Park, J.H.; Sohn, H.; Park, J.M. Synergistic effect of potassium metal siliconate on silicate conversion coating for corrosion protection of galvanized steel. J. Ind. Eng. Chem. 2012, 18, 655–660. [Google Scholar] [CrossRef]
- Rao, A.P.; Rao, A.V.; Pajonk, G.M. Hydrophobic and physical properties of the ambient pressure dried silica aerogels with sodium silicate precursor using various surface modification agents. Appl. Surf. Sci. 2007, 253, 6032–6040. [Google Scholar] [CrossRef]
- Pawlicka, A.; Tavares, F.C.; Dörr, D.S.; Cholant, C.M.; Ely, F.; Santos, M.J.L.; Avellaneda, C.O. Dielectric behavior and FTIR studies of xanthan gum-based solid polymer electrolytes. Electrochim. Acta 2019, 305, 232–239. [Google Scholar] [CrossRef]
- Ayeldeen, M.K.; Negm, A.M.; El Sawwaf, M.A. Evaluating the physical characteristics of biopolymer/soil mixtures. Arab. J. Geosci. 2016, 9, 371. [Google Scholar] [CrossRef]
- Muguda, S.; Booth, S.J.; Hughes, P.N.; Augarde, C.E.; Perlot, C.; Bruno, A.W.; Gallipoli, D. Mechanical properties of biopolymer-stabilised soil-based construction materials. Géotechnique Lett. 2017, 7, 309–314. [Google Scholar] [CrossRef]
- Guo, Z.; Zhu, Q.; Liu, C.; Xing, Z. Preparation of Ca-Al-Fe deicing salt and modified with sodium methyl silicate for reducing the influence of concrete structure. Constr. Build. Mater. 2018, 172, 263–271. [Google Scholar] [CrossRef]
- Chang, I.; Im, J.; Cho, G.C. Geotechnical engineering behaviors of gellan gum biopolymer treated sand. Can. Geotech. J. 2016, 53, 1658–1670. [Google Scholar] [CrossRef]
- Tingle, J.S.; Santoni, R.L. TRB, TRB, Stabilization of clay soils with nontraditional additives. In Proceedings of the 8th International Conference on Low Volume Roads, Reno, NV, USA, 22–25 June 2003; pp. A72–A84. [Google Scholar]
- Knox, A.S.; Petrisor, I.G.; Turick, C.E.; Roberts, J.; Paller, M.H.; Reible, D.D.; Forrest, C.R. Life span of biopolymer sequestering agents for contaminant removal and erosion resistance. Biopolymers 2010, 81–108. [Google Scholar] [CrossRef]
- Etemadi, O.; Petrisor, I.G.; Kim, D.; Wan, M.; Yen, T.F. Stabilization of Metals in Subsurface by Biopolymers: Laboratory Drainage Flow Studies. Soil Sediment Contam. Int. J. 2003, 12, 647–661. [Google Scholar] [CrossRef]
- Wu, F.; Chen, B.; Qu, G.; Liu, S.; Zhao, C.; Ren, Y.; Liu, X. Harmless treatment technology of phosphogypsum: Directional stabilization of toxic and harmful substances. J. Environ. Manag. 2022, 311, 114827. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition | SO3 | CaO | SiO2 | Al2O3 | P2O5 | Fe2O3 | F− | Cl− | Cr2O3 |
|---|---|---|---|---|---|---|---|---|---|
| Component (wt.%) | 43.47 | 28.79 | 4.72 | 0.877 | 0.615 | 0.358 | 0. 14 | 0.0060 | 0.0026 |
| Method Parameters | |
|---|---|
| eluent type | KOH |
| eluent flow (L/min) | 1.0 |
| column | AS19 |
| Temperature (°C) | 30 |
| suppressor | AERS-4 mm |
| Current (mA) | 50 |
| detector | Conductivity detector |
| injection volume | 25 μL |
| Method Parameters | |
|---|---|
| Plasma gas flow (L/min) | 0.5 |
| Nebulizer and auxiliary gas flow (L/min) | 1.50 |
| RF Power (kW) | 1.55 |
| Integration time (s) | 0.9 |
| Replicates per sample | 3 |
| Mode of operation | He |
| Cr/(μg·L−1) | F−/(mg·L−1) | PO43−/(mg·L−1) | |
|---|---|---|---|
| PG | 1.807 | 15.784 | 23.731 |
| 1.0% XG | 0.462 | 2.533 | 9.939 |
| 2.0% XG | 0.449 | 2.821 | 8.297 |
| 3.0% XG | 0.458 | 2.780 | 9.202 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Xu, T.; Chu, H.; Xi, X.; Zhang, F.; Jin, W. Study on Synergistic Effect of Xanthan Gum and Sodium Methylsiliconate on Mechanical Strength and Water Stability of Phosphogypsum Road-Based Materials. Materials 2023, 16, 6766. https://doi.org/10.3390/ma16206766
Wu J, Xu T, Chu H, Xi X, Zhang F, Jin W. Study on Synergistic Effect of Xanthan Gum and Sodium Methylsiliconate on Mechanical Strength and Water Stability of Phosphogypsum Road-Based Materials. Materials. 2023; 16(20):6766. https://doi.org/10.3390/ma16206766
Chicago/Turabian StyleWu, Jianhui, Tong Xu, Hongqiang Chu, Xiang Xi, Fengchen Zhang, and Weizhun Jin. 2023. "Study on Synergistic Effect of Xanthan Gum and Sodium Methylsiliconate on Mechanical Strength and Water Stability of Phosphogypsum Road-Based Materials" Materials 16, no. 20: 6766. https://doi.org/10.3390/ma16206766
APA StyleWu, J., Xu, T., Chu, H., Xi, X., Zhang, F., & Jin, W. (2023). Study on Synergistic Effect of Xanthan Gum and Sodium Methylsiliconate on Mechanical Strength and Water Stability of Phosphogypsum Road-Based Materials. Materials, 16(20), 6766. https://doi.org/10.3390/ma16206766