Effect of Film Thickness on the Self-Assembly of CBABC Symmetric Pentablock Terpolymer Melts under 1D Confinement: A Dissipative Particle Dynamic Study

Abstract
:1. Introduction
2. Simulation Methods
3. Results and Discussion
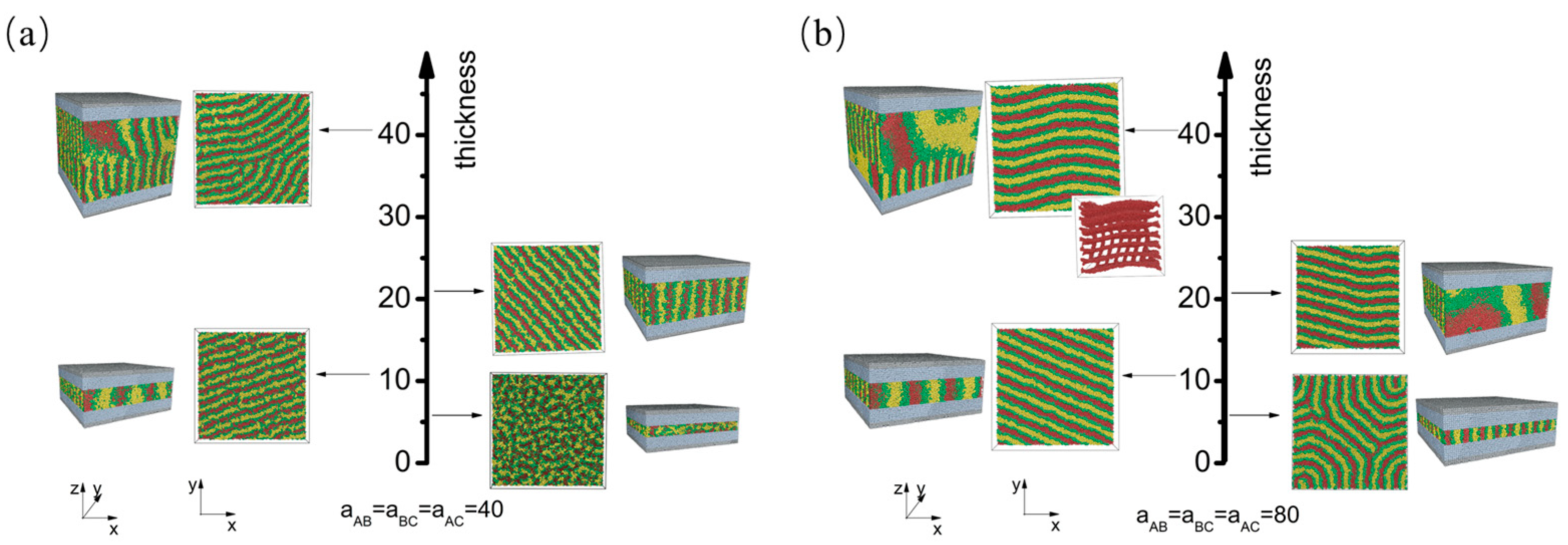
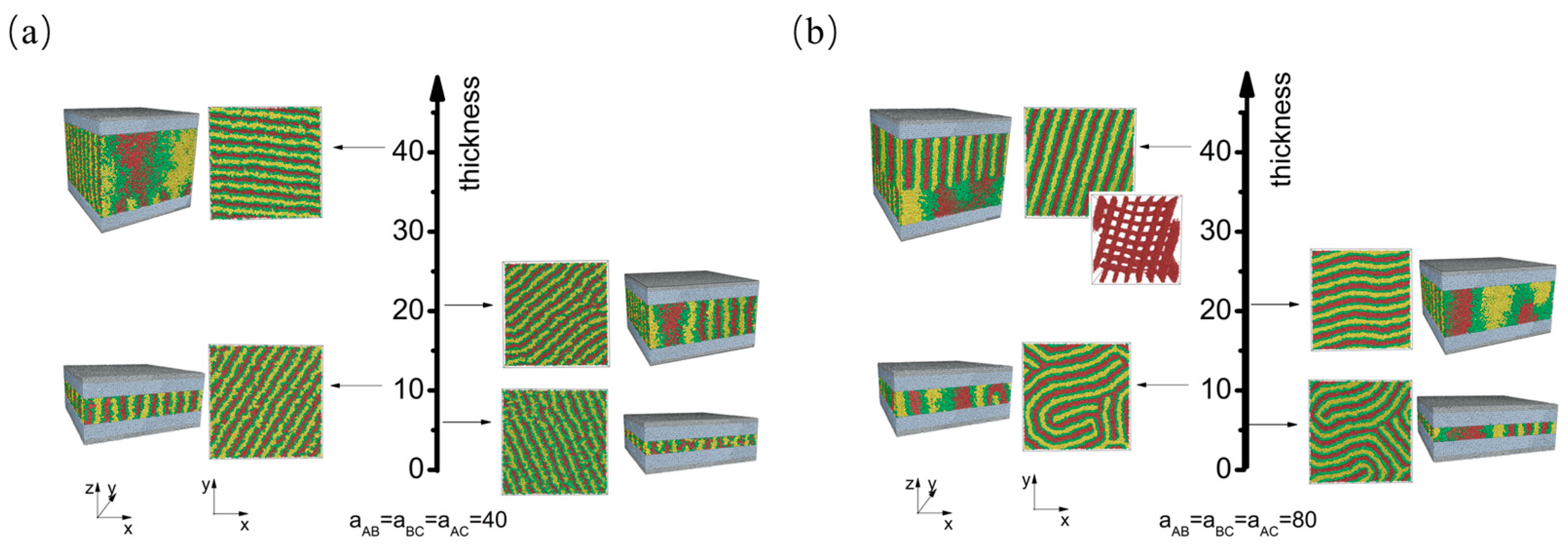
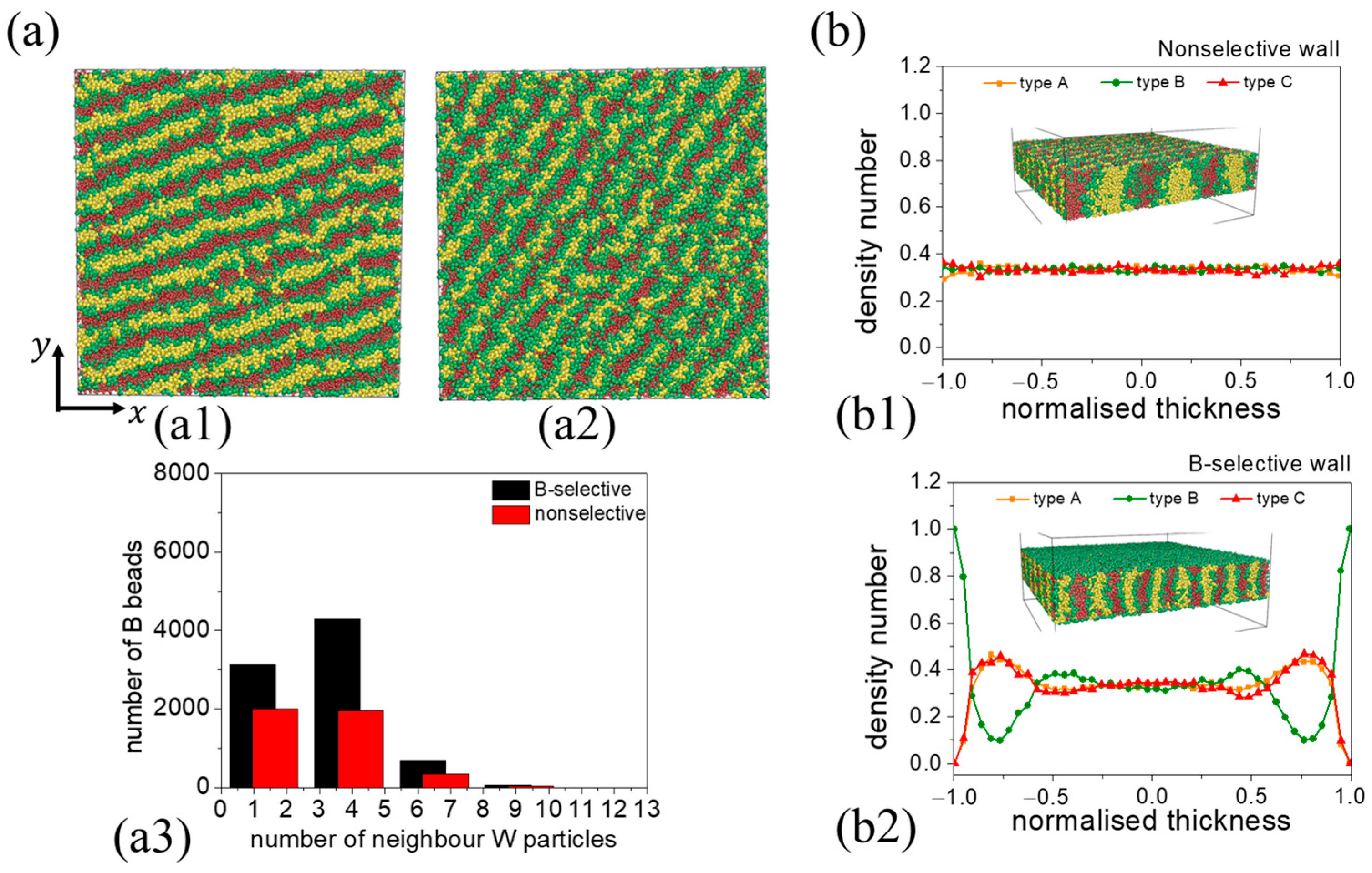
3.1. Nonselective Wall
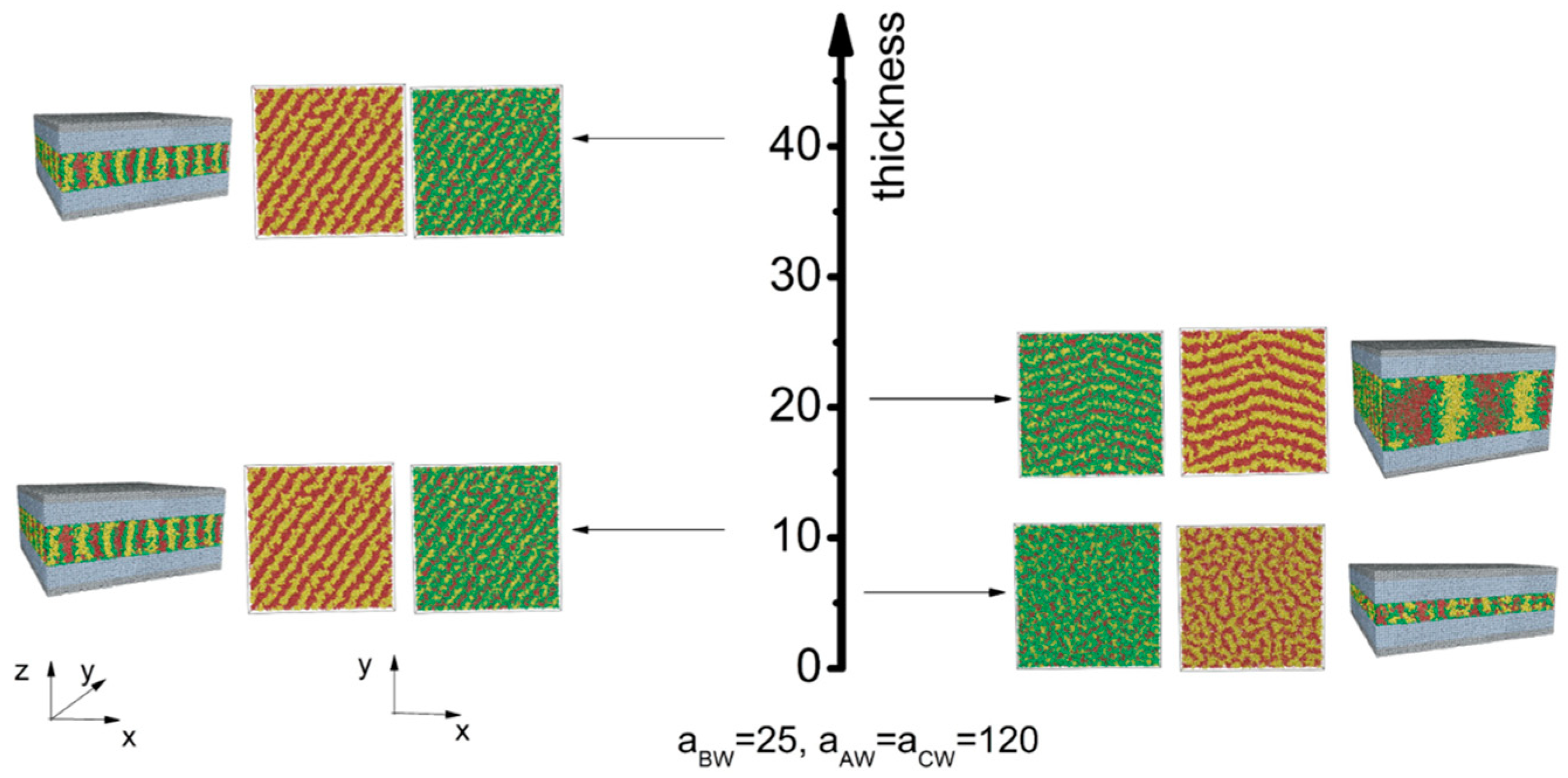
3.2. Selective Wall
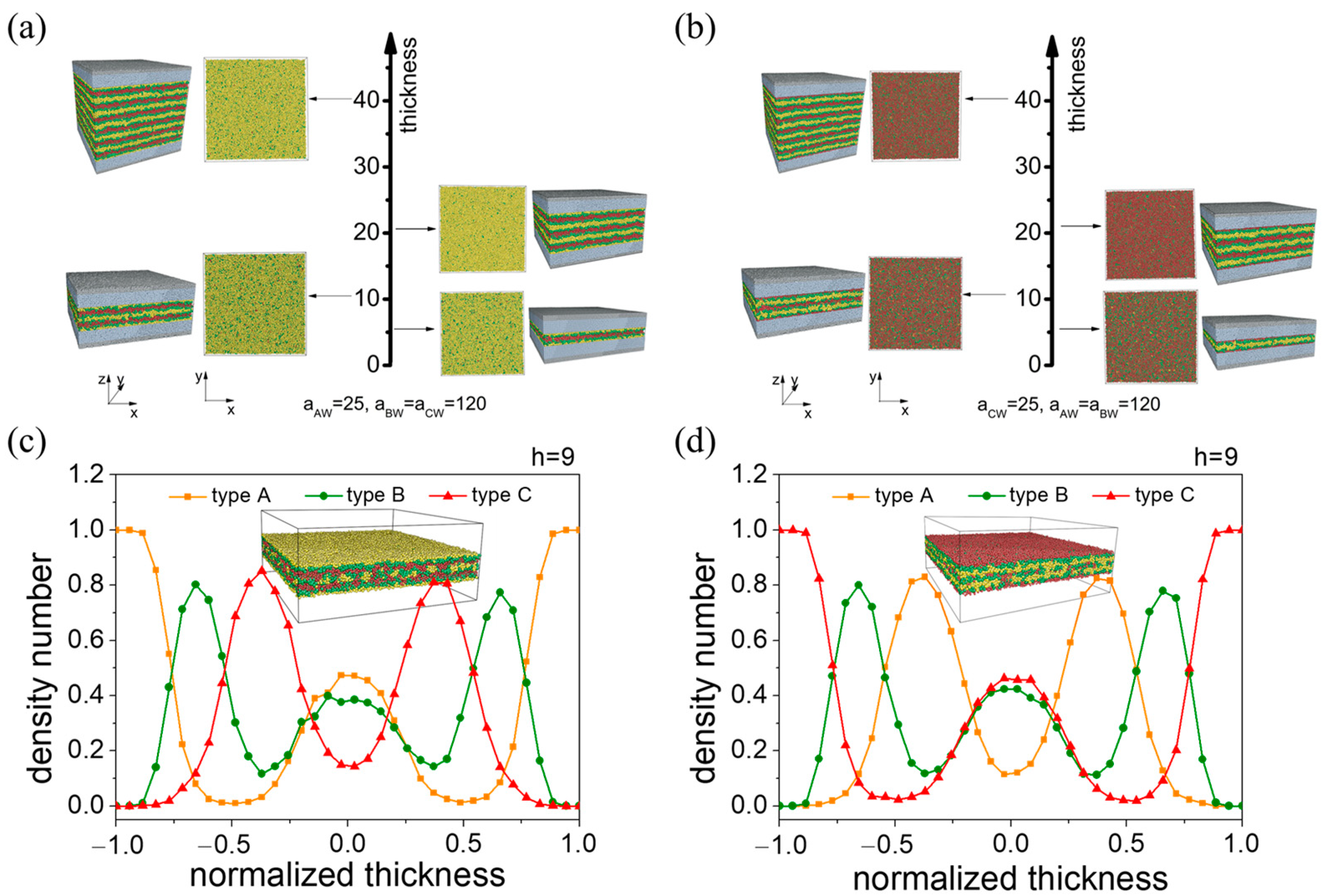
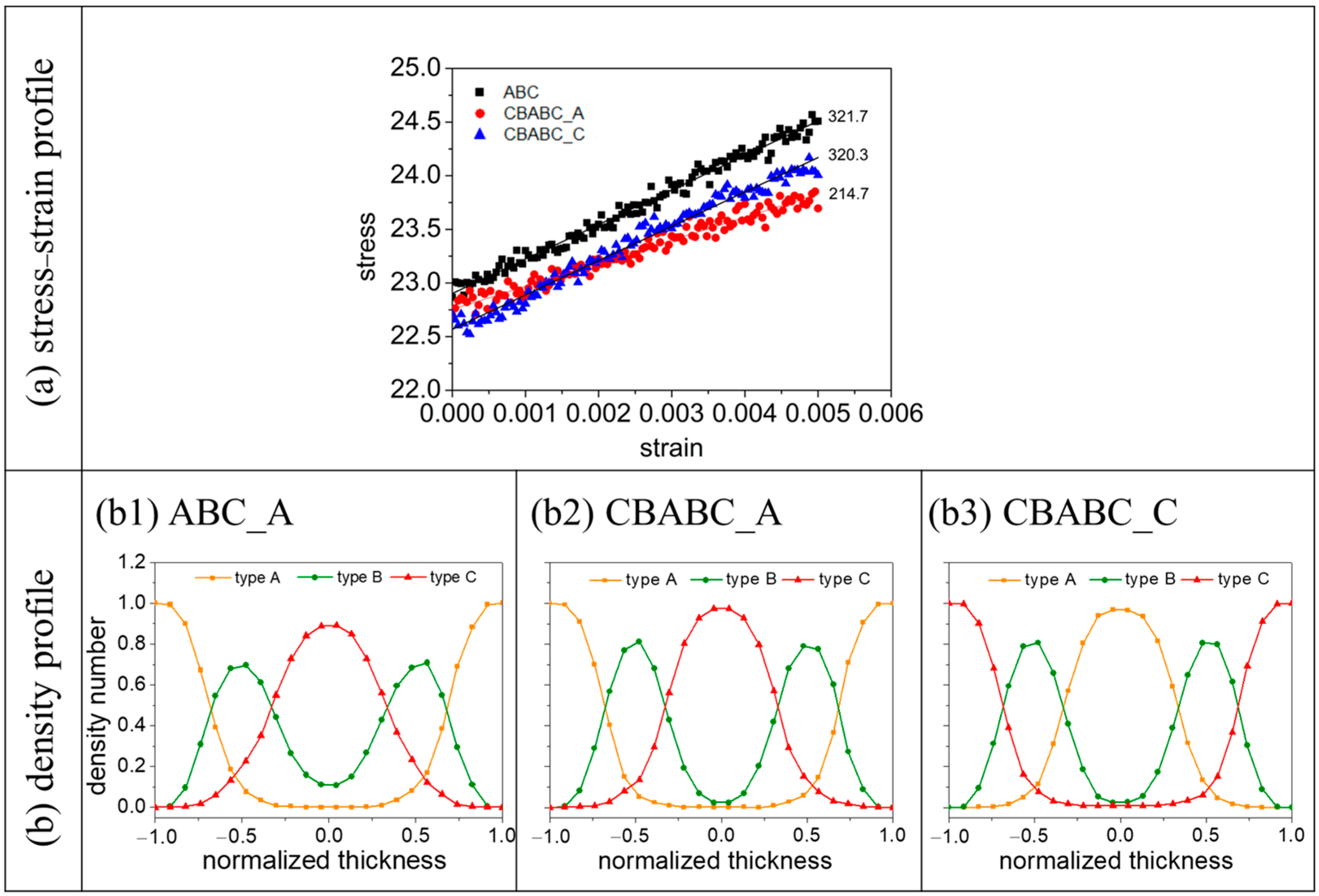
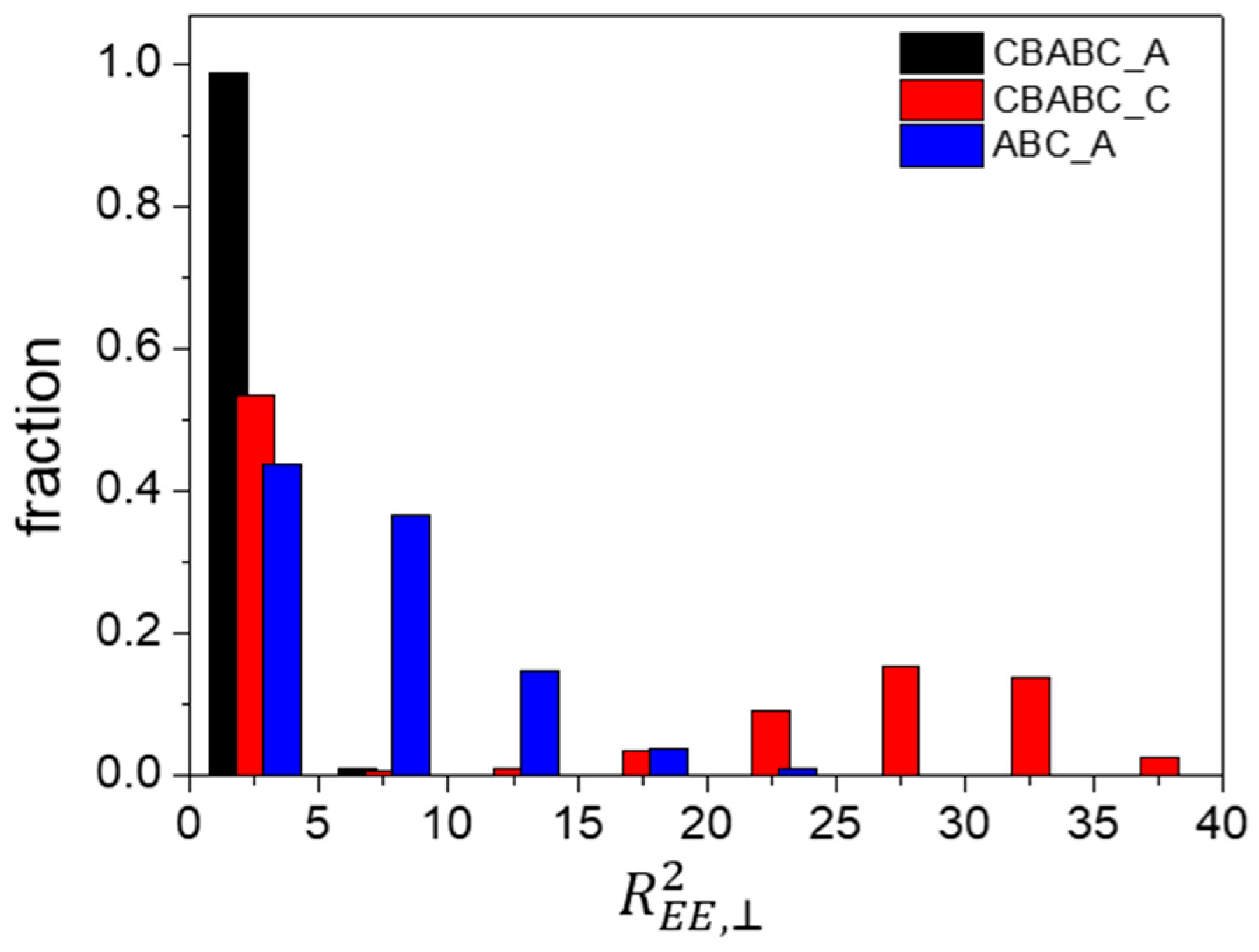
3.2.1. A-Block- and C-Block-Selective Wall
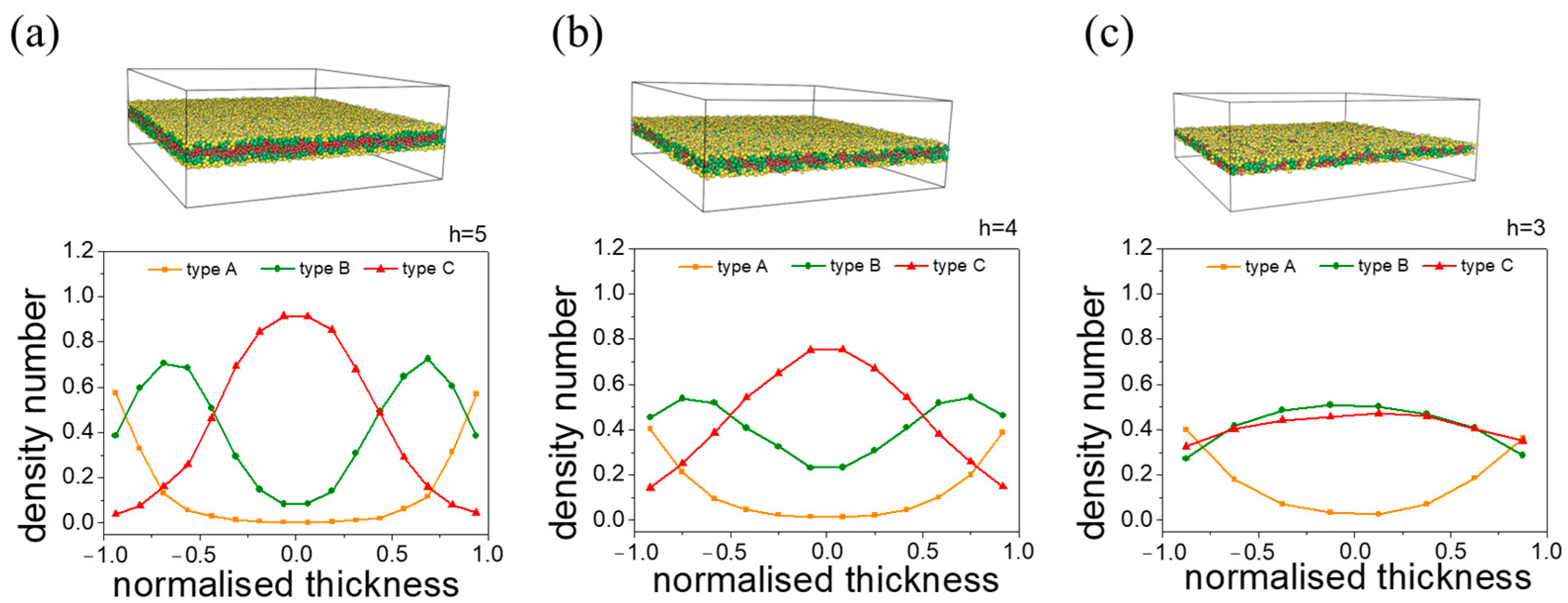
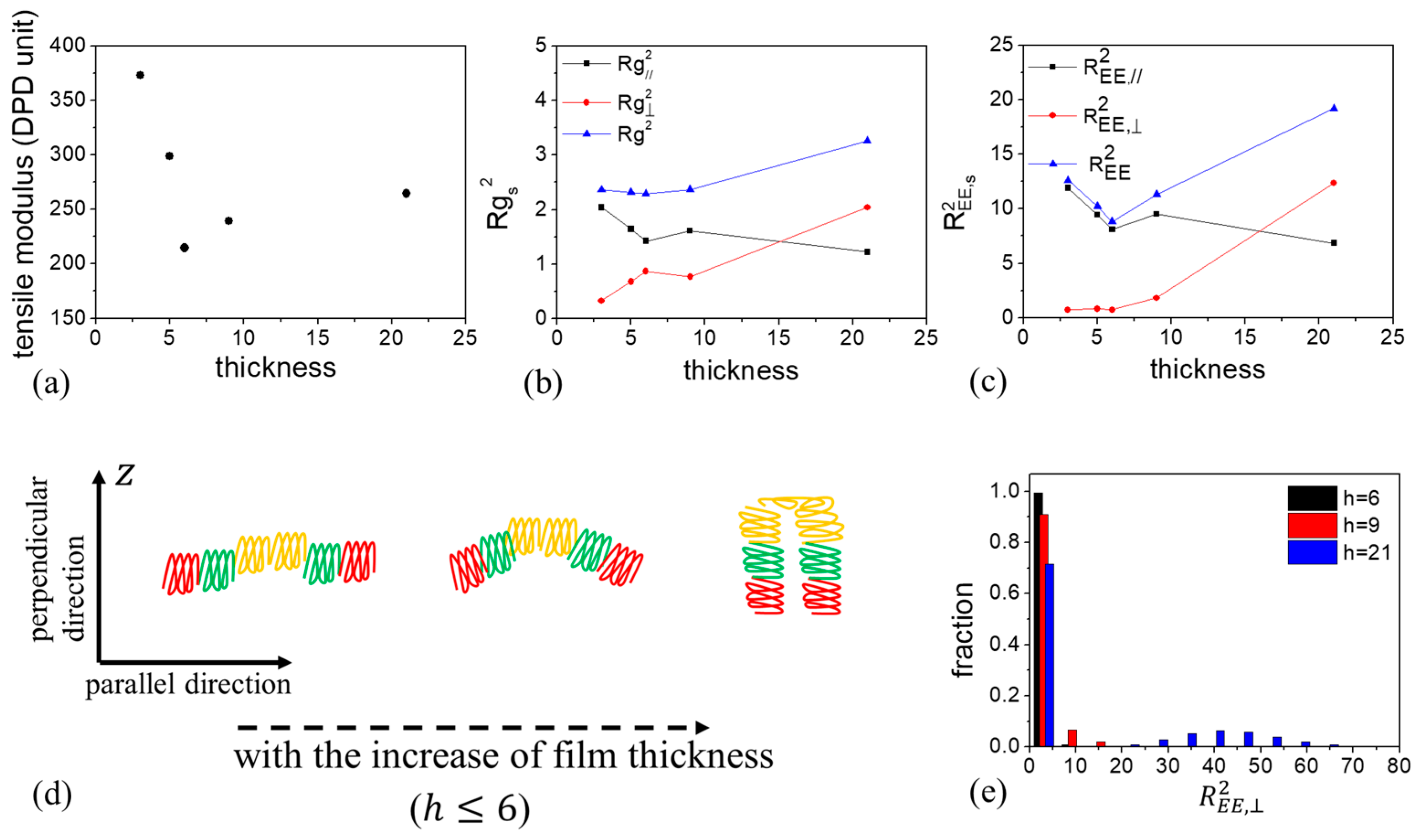
3.2.2. B-Block-Selective Wall
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shi, A.-C.; Li, B. Self-assembly of diblock copolymers under confinement. Soft Matter 2012, 9, 1398–1413. [Google Scholar] [CrossRef]
- Hu, X.-H.; Xiong, S. Fabrication of Nanodevices Through Block Copolymer Self-Assembly. Front. Nanotechnol. 2022, 4, 762996. [Google Scholar] [CrossRef]
- Beyer, V.P.; Kim, J.; Becer, C.R. Synthetic approaches for multiblock copolymers. Polym. Chem. 2020, 11, 1271–1291. [Google Scholar] [CrossRef]
- De Neve, J.; Haven, J.J.; Maes, L.; Junkers, T. Sequence-definition from controlled polymerization: The next generation of materials. Polym. Chem. 2018, 9, 4692–4705. [Google Scholar] [CrossRef]
- Lutz, J.-F. Writing on Polymer Chains. Acc. Chem. Res. 2013, 46, 2696–2705. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Xu, J.; Du, B. Self-assembly of ABCBA Linear Pentablock Terpolymers. Polym. Rev. 2023, 1–35. [Google Scholar] [CrossRef]
- Gao, J.; Lv, C.; An, K.; Gu, X.; Nie, J.; Li, Y.; Xu, J.; Du, B. Observation of Double Gyroid and Hexagonally Perforated Lamellar Phases in ABCBA Pentablock Terpolymers. Macromolecules 2020, 53, 9641–9653. [Google Scholar] [CrossRef]
- Kopchick, J.G.; Storey, R.F.; Beyer, F.L.; Mauritz, K.A. Poly[acrylic acid-b-styrene-b-isobutylene-b-styrene-b-acrylic acid] pentablock terpolymers: 1. Morphological characterization. Polymer 2007, 48, 3739–3748. [Google Scholar] [CrossRef]
- Kopchick, J.G.; Storey, R.F.; Jarrett, W.L.; Mauritz, K.A. Morphology of poly[(t-butyl acrylate)-b-styrene-b-isobutylene-b-styrene-b-(t-butyl acrylate)] pentablock terpolymers and their thermal conversion to the acrylic acid form. Polymer 2008, 49, 5045–5052. [Google Scholar] [CrossRef]
- Meuler, A.J.; Fleury, G.; Hillmyer, M.A.; Bates, F.S. Structure and Mechanical Properties of an O70 (Fddd) Network-Forming Pentablock Terpolymer. Macromolecules 2008, 41, 5809–5817. [Google Scholar] [CrossRef]
- Nishiwaki, Y.; Masutani, K.; Kimura, Y.; Lee, C.-W. Synthesis and mechanochemical properties of biobased ABCBA-type pentablock copolymers comprising poly-d-lactide (A), poly-l-lactide (B) and poly(1,2-propylene succinate) (C). J. Polym. Sci. 2022, 60, 2043–2054. [Google Scholar] [CrossRef]
- Liu, H.-H.; Huang, C.-I.; Shi, A.-C. Self-Assembly of Linear ABCBA Pentablock Terpolymers. Macromolecules 2015, 48, 6214–6223. [Google Scholar] [CrossRef]
- Lo, Y.; Chang, C.; Liu, H.; Huang, C.; Shi, A. Self-Assembly of Nonfrustrated ABCBA Linear Pentablock Terpolymers. Macromol. Theory Simul. 2021, 30, 2100014. [Google Scholar] [CrossRef]
- Huinink, H.P.; Brokken-Zijp, J.C.M.; van Dijk, M.A.; Sevink, G.J.A. Asymmetric block copolymers confined in a thin film. J. Chem. Phys. 2000, 112, 2452–2462. [Google Scholar] [CrossRef]
- Matsen, M.W. Thin films of block copolymer. J. Chem. Phys. 1997, 106, 7781–7791. [Google Scholar] [CrossRef]
- Epps, T.H.; DeLongchamp, D.M.; Fasolka, M.J.; Fischer, D.A.; Jablonski, E.L. Substrate Surface Energy Dependent Morphology and Dewetting in an ABC Triblock Copolymer Film. Langmuir 2007, 23, 3355–3362. [Google Scholar] [CrossRef]
- Albert, J.N.L.; Epps, T.H. Self-assembly of block copolymer thin films. Mater. Today 2010, 13, 24–33. [Google Scholar] [CrossRef]
- Hu, H.; Gopinadhan, M.; Osuji, C.O. Directed self-assembly of block copolymers: A tutorial review of strategies for enabling nanotechnology with soft matter. Soft Matter 2014, 10, 3867–3889. [Google Scholar] [CrossRef]
- Kim, D.H.; Suh, A.; Park, G.; Yoon, D.K.; Kim, S.Y. Nanoscratch-Directed Self-Assembly of Block Copolymer Thin Films. ACS Appl. Mater. Interfaces 2021, 13, 5772–5781. [Google Scholar] [CrossRef]
- Li, W.; Liu, M.; Qiu, F.; Shi, A.-C. Phase Diagram of Diblock Copolymers Confined in Thin Films. J. Phys. Chem. B 2013, 117, 5280–5288. [Google Scholar] [CrossRef]
- Mu, D.; Li, J.-Q.; Feng, S.-Y. One-dimensional Confinement Effect on the Self-assembly of Symmetric H-shaped Copolymers in a Thin Film. Sci. Rep. 2017, 7, 13610. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.-W.; Chang, J.-H.; Chang, I.C.-Y.; Sun, Y.-S. Phase behavior in thin films of weakly segregated block copolymer/homopolymer blends. Soft Matter 2021, 17, 9189–9197. [Google Scholar] [CrossRef] [PubMed]
- Aviv, Y.; Altay, E.; Burg, O.; Müller, M.; Rzayev, J.; Shenhar, R. Bottlebrush Block Copolymer Assembly in Ultraconfined Films: Effect of Substrate Selectivity. Macromolecules 2021, 54, 2079–2089. [Google Scholar] [CrossRef]
- Michman, E.; Langenberg, M.; Stenger, R.; Oded, M.; Schvartzman, M.; Mueller, M.; Shenhar, R. Controlled Spacing between Nanopatterned Regions in Block Copolymer Films Obtained by Utilizing Substrate Topography for Local Film Thickness Differentiation. ACS Appl. Mater. Interfaces 2019, 11, 35247–35254. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Huh, J.; Carter, K.R.; Russell, T.P. Directed Self-Assembly of Block Copolymer Thin Films Using Minimal Topographic Patterns. ACS Nano. 2016, 10, 7915–7925. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-W.; Kim, E.; Lee, H.; Berry, B.C.; Kim, H.-C.; Ryu, D.Y. Thickness-dependent ordering of perpendicularly oriented lamellae in PS-b-PMMA thin films. Polymer 2015, 74, 63–69. [Google Scholar] [CrossRef]
- Diaz, J.; Pinna, M.; Breen, C.; Zvelindovsky, A.; Pagonabarraga, I. Block Copolymer Nanocomposites under Confinement: Effect on Frustrated Phases. Macromolecules 2023, 56, 5010–5021. [Google Scholar] [CrossRef]
- Mu, D.; Li, J.-Q.; Cong, X.-S.; Zhang, H. Mesoscopic Detection of the Influence of a Third Component on the Self-Assembly Structure of A2B Star Copolymer in Thin Films. Polymers 2019, 11, 1636. [Google Scholar] [CrossRef]
- Spencer, R.K.; Matsen, M.W. Confinement effects on the miscibility of block copolymer blends. Eur. Phys. J. E 2016, 39, 43. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Zhang, L.; Lin, J.; Jiang, T. Controllable Hierarchical Microstructures Self-Assembled from Multiblock Copolymers Confined in Thin Films. Langmuir 2015, 31, 2533–2544. [Google Scholar] [CrossRef]
- Petrus, P.; Lísal, M.; Brennan, J.K. Self-Assembly of Symmetric Diblock Copolymers in Planar Slits with and without Nanopatterns: Insight from Dissipative Particle Dynamics Simulations. Langmuir 2009, 26, 3695–3709. [Google Scholar] [CrossRef] [PubMed]
- Groot, R.D.; Madden, T.J. Dynamic simulation of diblock copolymer microphase separation. J. Chem. Phys. 1998, 108, 8713–8724. [Google Scholar] [CrossRef]
- Yu, B.; Li, B.; Jin, Q.; Ding, D.; Shi, A.-C. Confined self-assembly of cylinder-forming diblock copolymers: Effects of confining geometries. Soft Matter 2011, 7, 10227–10240. [Google Scholar] [CrossRef]
- Yong, D.; Kim, Y.; Jo, S.; Ryu, D.Y.; Kim, J.U. Order-to-Disorder Transition of Cylinder-Forming Block Copolymer Films Confined within Neutral Interfaces. Macromolecules 2021, 54, 11304–11315. [Google Scholar] [CrossRef]
- Matsen, M.W.; Thompson, R.B. Equilibrium behavior of symmetric ABA triblock copolymer melts. J. Chem. Phys. 1999, 111, 7139–7146. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| aij (DPD Units) | A | B | C | W (Wall) |
|---|---|---|---|---|
| A | 25 | |||
| B | 40/80 | 25 | ||
| C | 40/80 | 40/80 | 25 | |
| W (wall) | 25/120 | 25/120 | 25/120 | 25 |
| Chain | ||||
|---|---|---|---|---|
| ABC_A | 1.64 | 1.02 | 2.66 | |
| CBABC_C | 1.37 | 1.84 | 3.21 | |
| CBABC_A | 1.42 | 0.87 | 2.29 | |
| Chain | ||||
|---|---|---|---|---|
| ABC_A | 9.67 | 6.43 | 16.09 | |
| CBABC_C | 7.91 | 12.77 | 20.68 | |
| CBABC_A | 8.10 | 0.73 | 8.83 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y. Effect of Film Thickness on the Self-Assembly of CBABC Symmetric Pentablock Terpolymer Melts under 1D Confinement: A Dissipative Particle Dynamic Study. Materials 2023, 16, 6862. https://doi.org/10.3390/ma16216862
Guo Y. Effect of Film Thickness on the Self-Assembly of CBABC Symmetric Pentablock Terpolymer Melts under 1D Confinement: A Dissipative Particle Dynamic Study. Materials. 2023; 16(21):6862. https://doi.org/10.3390/ma16216862
Chicago/Turabian StyleGuo, Yingying. 2023. "Effect of Film Thickness on the Self-Assembly of CBABC Symmetric Pentablock Terpolymer Melts under 1D Confinement: A Dissipative Particle Dynamic Study" Materials 16, no. 21: 6862. https://doi.org/10.3390/ma16216862
APA StyleGuo, Y. (2023). Effect of Film Thickness on the Self-Assembly of CBABC Symmetric Pentablock Terpolymer Melts under 1D Confinement: A Dissipative Particle Dynamic Study. Materials, 16(21), 6862. https://doi.org/10.3390/ma16216862