
Improved Dimethyl Ether Production from Syngas over Aerogel Sulfated Zirconia and Cu-ZnO(Al) Bifunctional Composite Catalysts

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Catalyst Preparation
2.1.1. Preparation of Sulfated Zirconia (ZrO2/SO42) Acid Catalysts
2.1.2. Preparation of Cu-ZnO-Al (CZA) Catalyst
2.1.3. Preparation of CZA-ZrO2/SO4 Bifunctional Composite Catalysts
2.1.4. Catalysts Characterization
2.2. Catalytic Activity in the Direct Synthesis of DME from Syngas
- CO conversion:
- CO2 selectivity:
- DME selectivity:
- CH3OH selectivity:
- DME production rate:
- CH3OH production rate:where M refers to the molar flow rate of each product.
3. Results
3.1. Characterization of Sulfated Zirconia Acid Catalysts
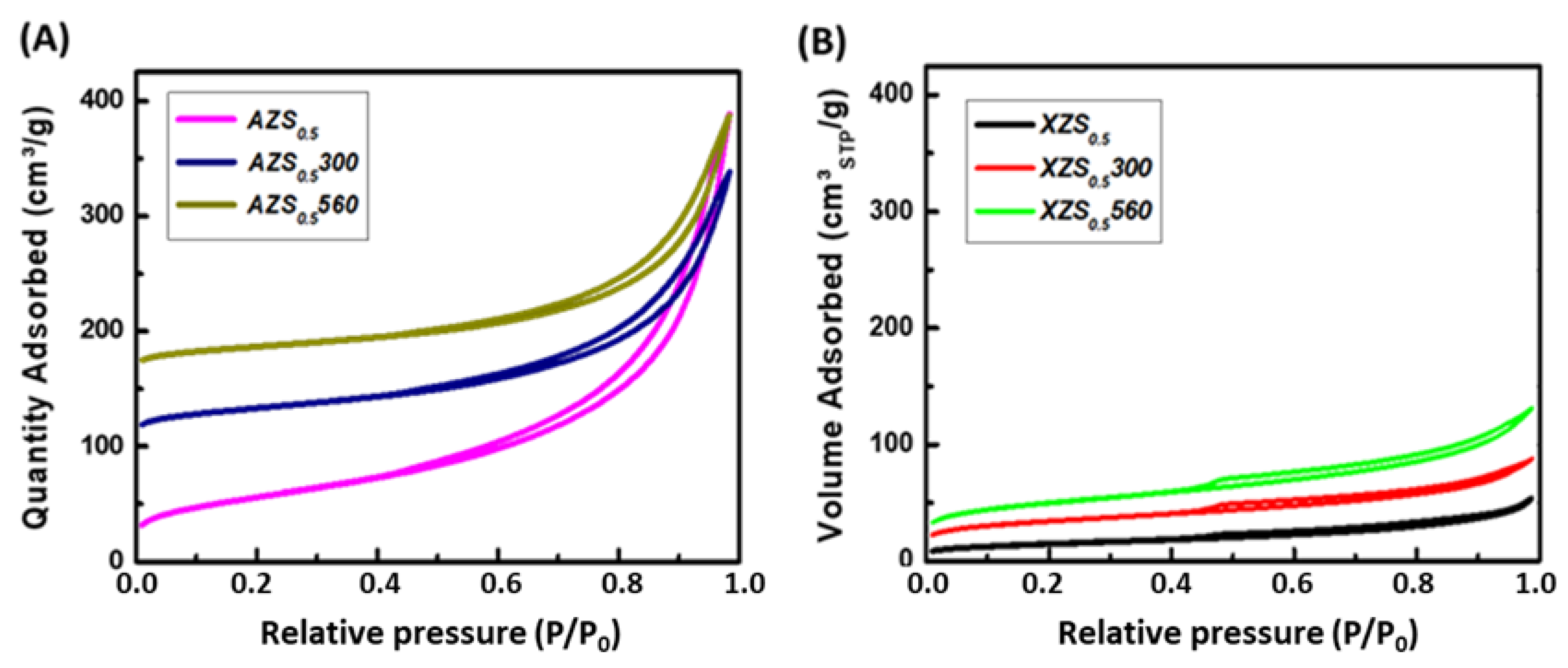
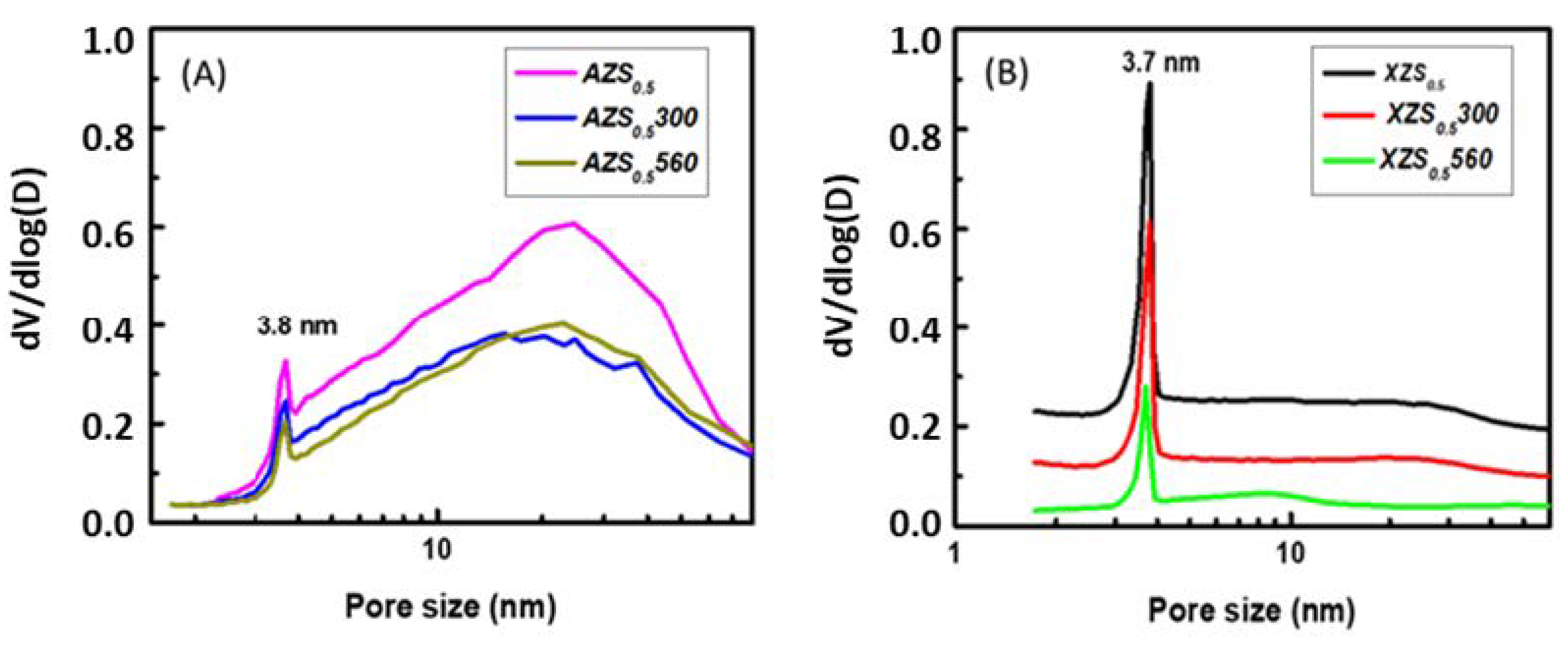
3.1.1. Textural Properties
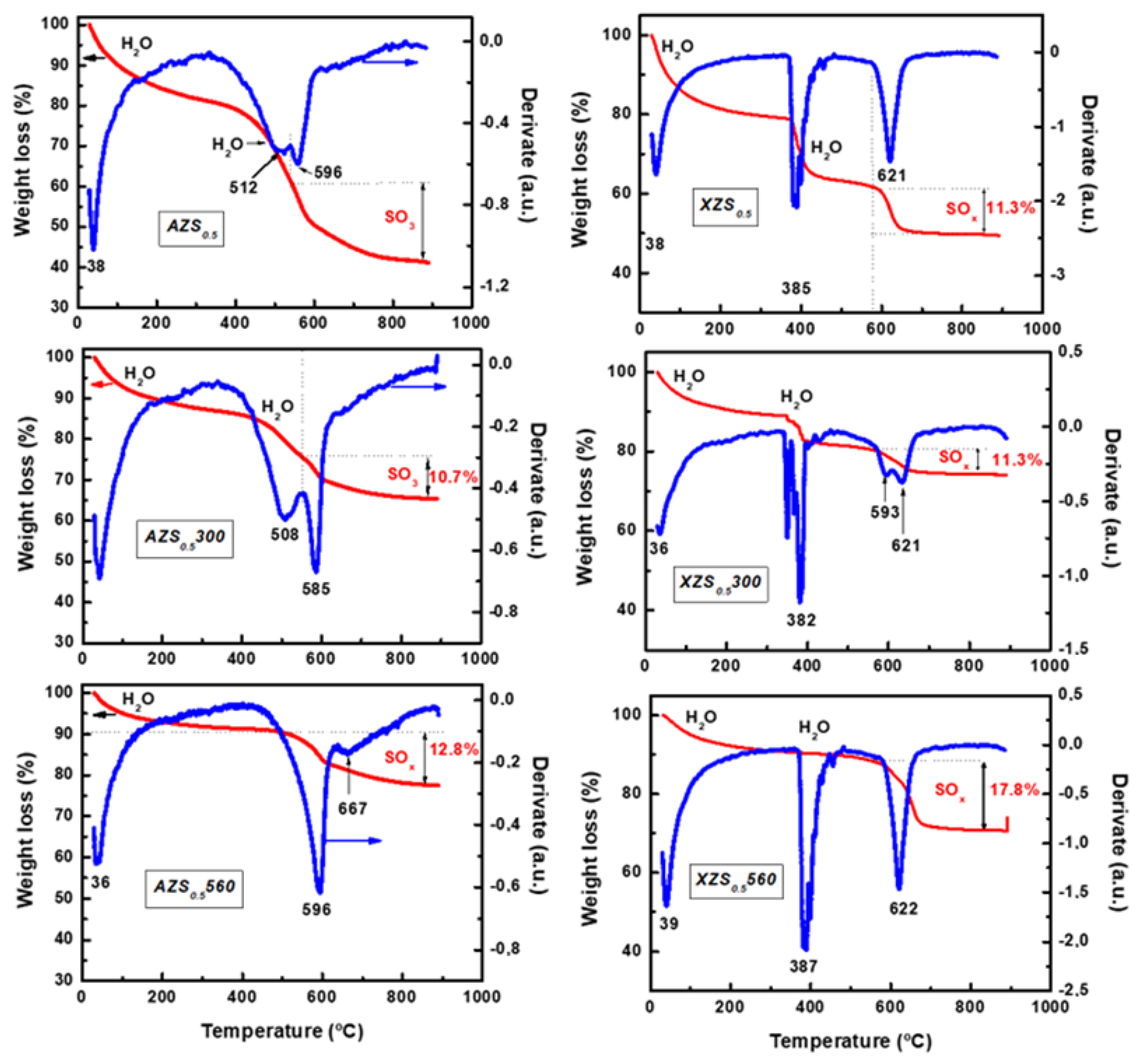
3.1.2. TGA/DTA
3.1.3. XRD Characterization
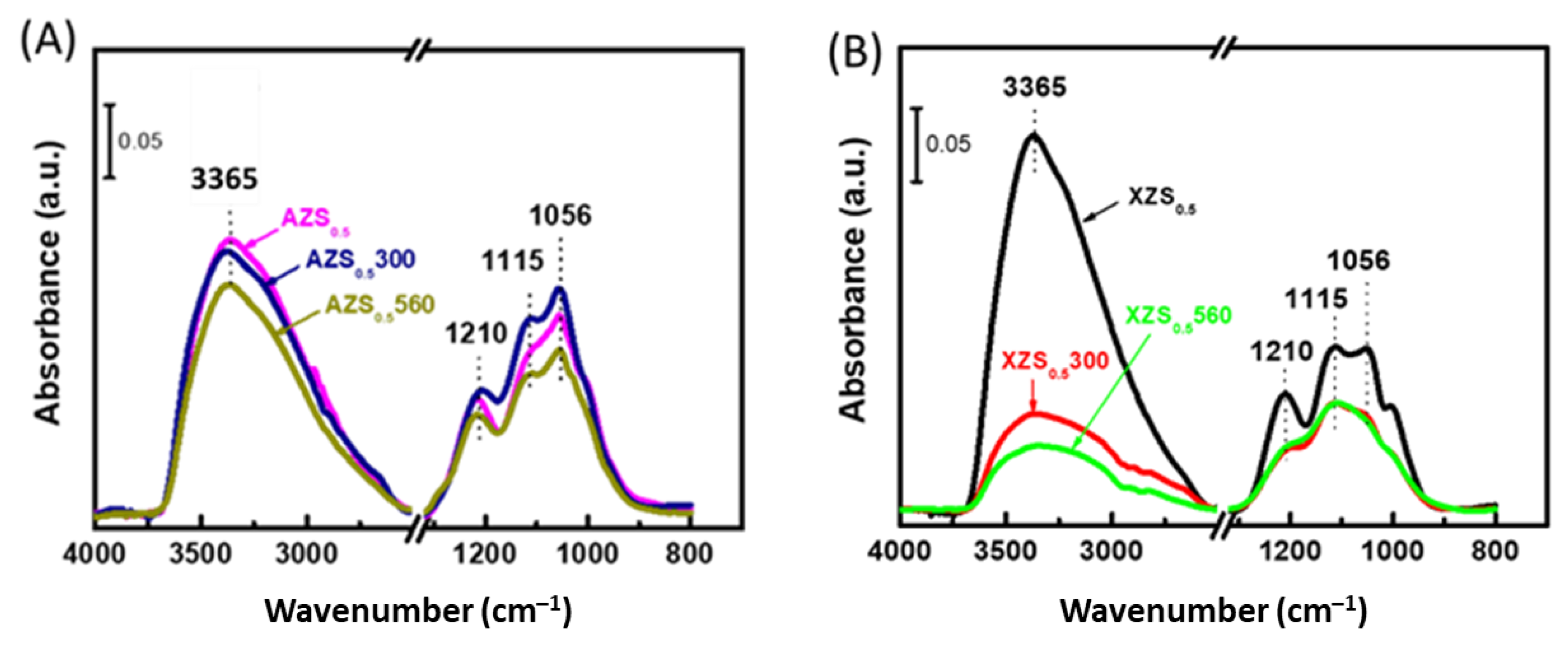
3.1.4. FTIR Study
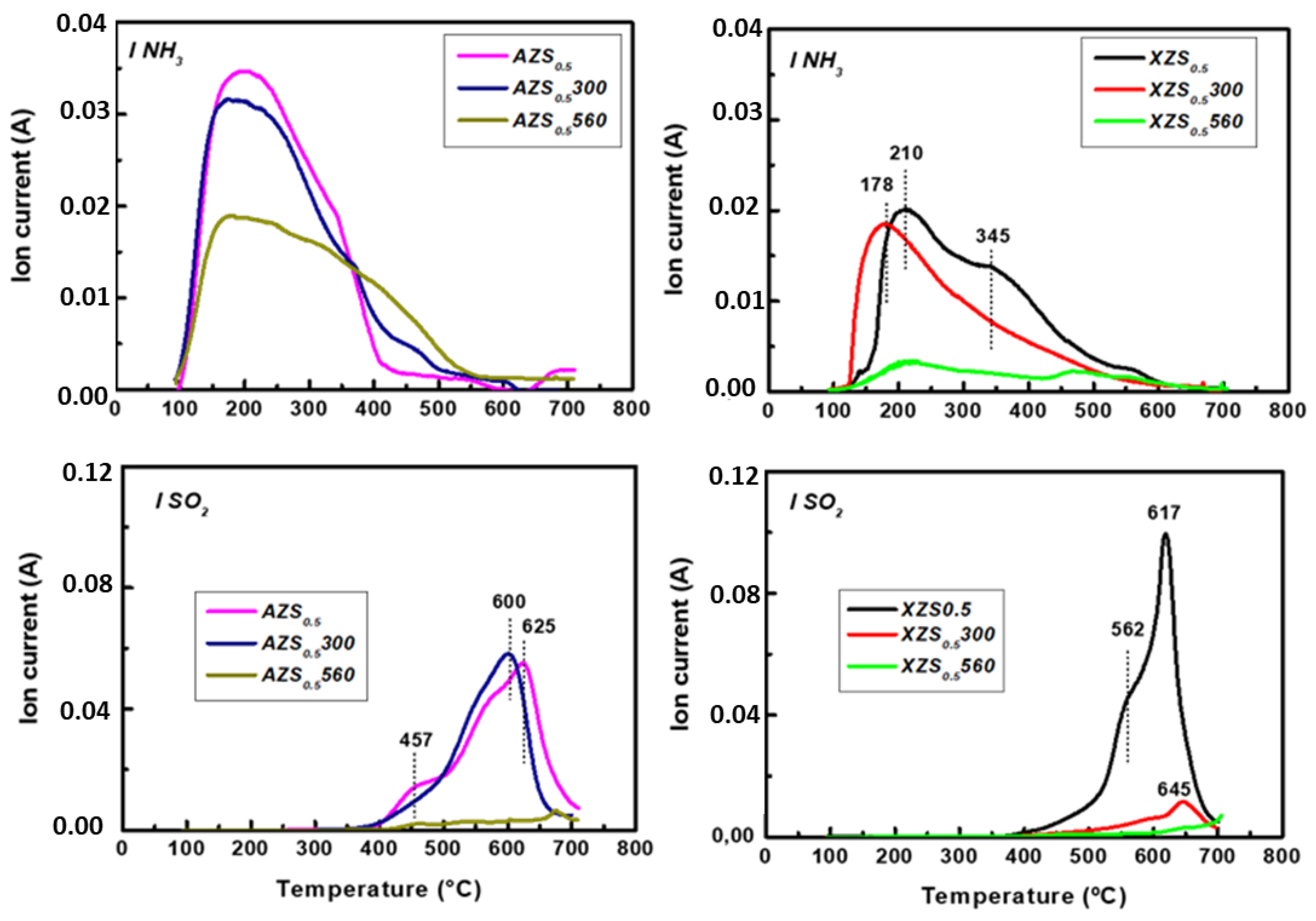
3.1.5. TPD/MS of NH3
3.2. Characterization of CZA-ZrO2/SO4 Bifunctional Composite Catalysts
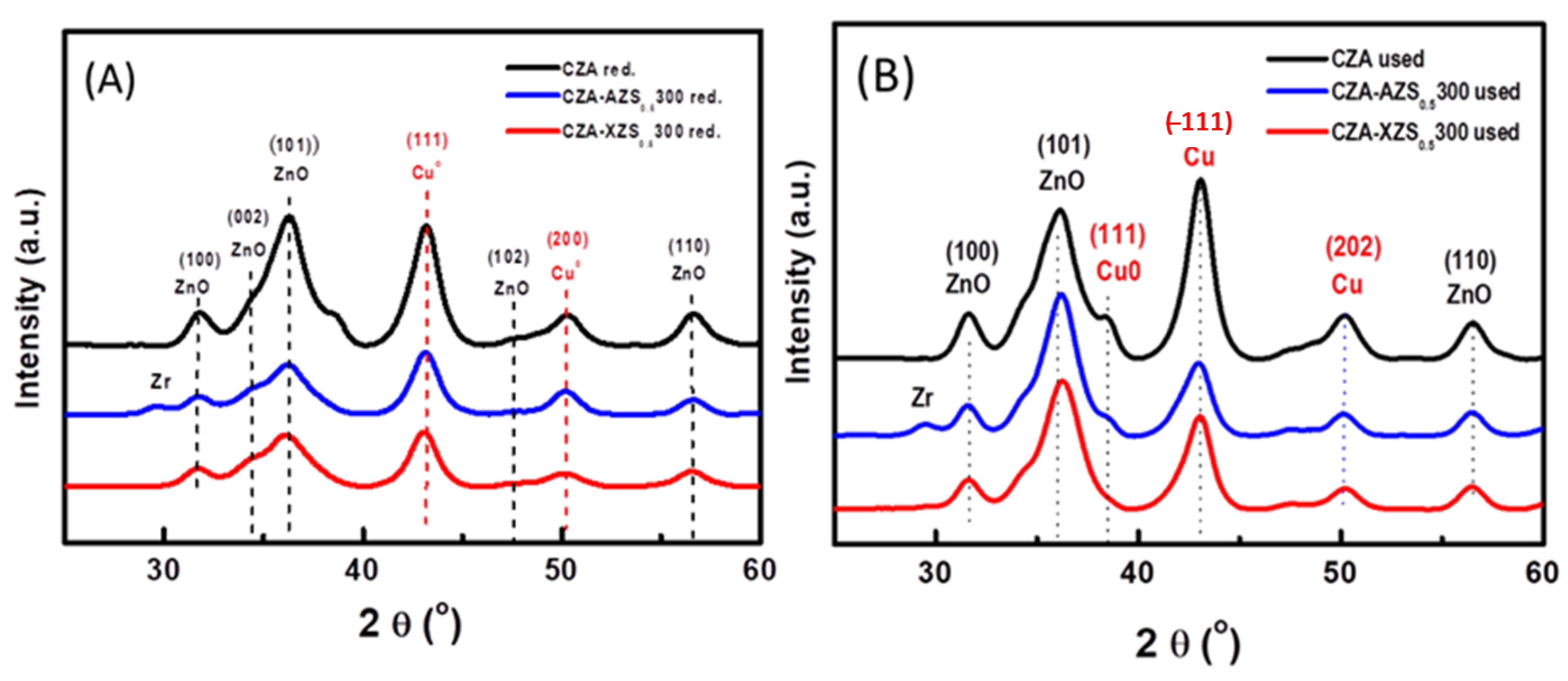
3.2.1. XRD Characterization of Bifunctional Calcined Catalysts
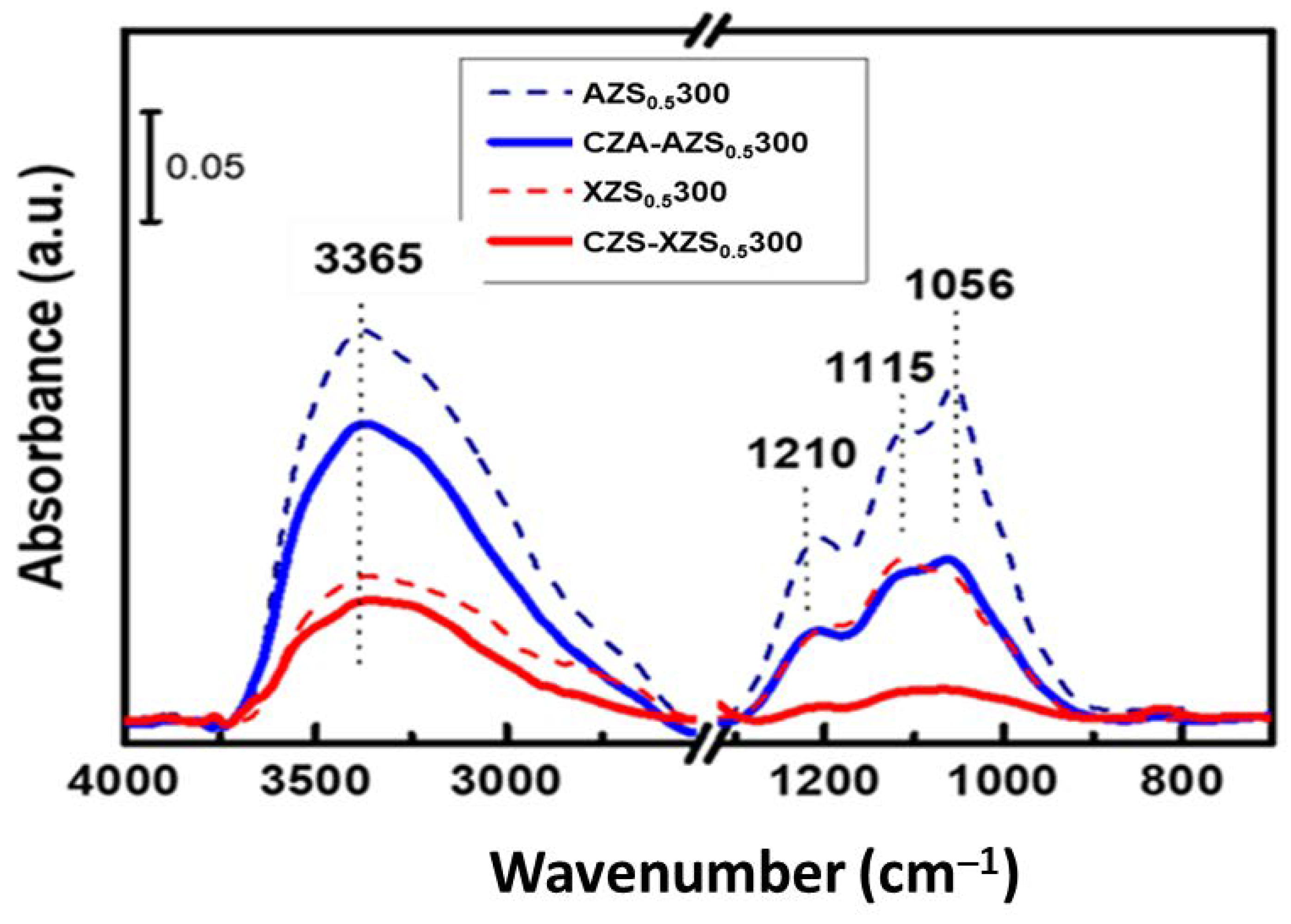
3.2.2. FTIR of Bifunctional Calcined Catalysts
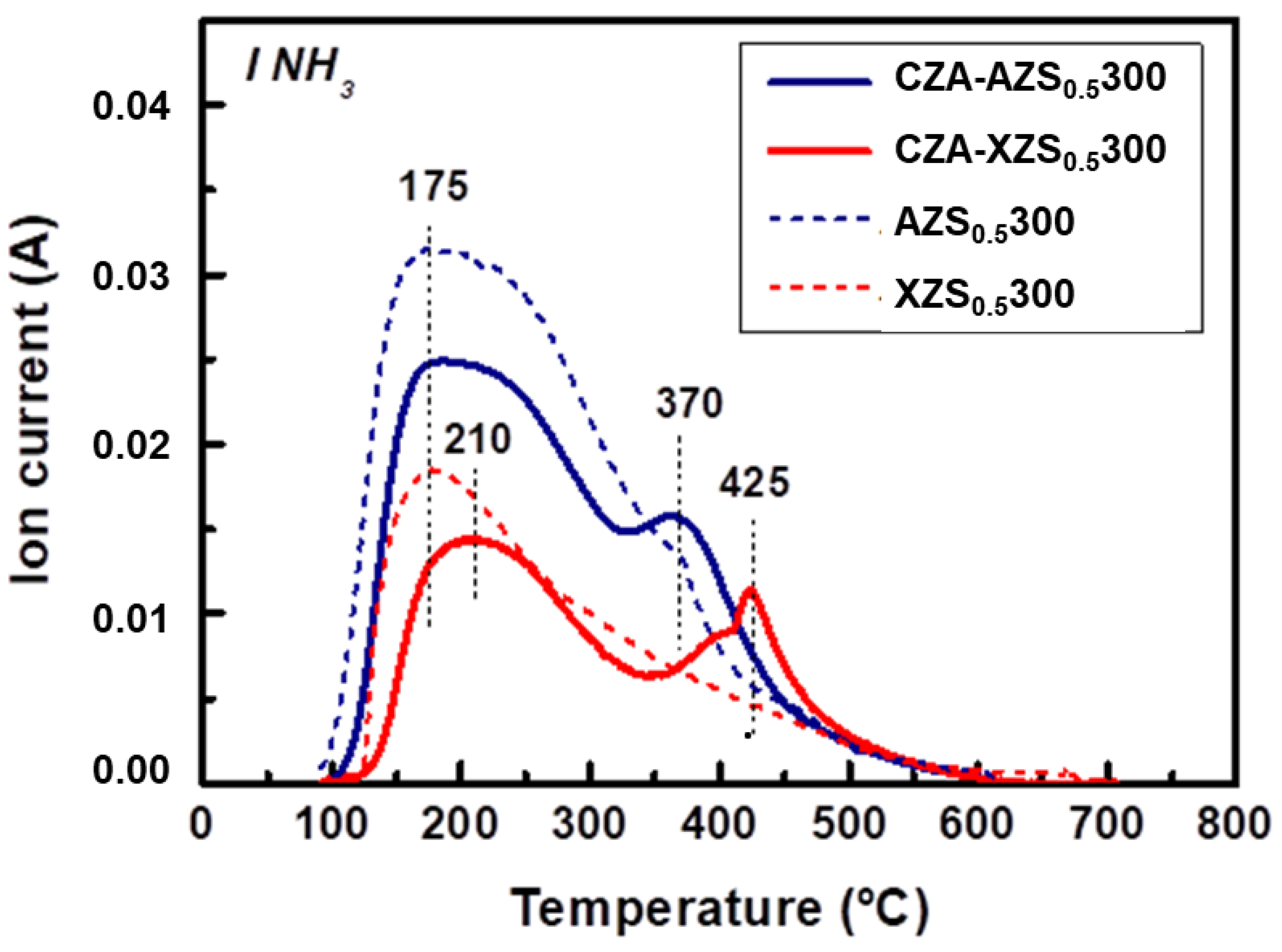
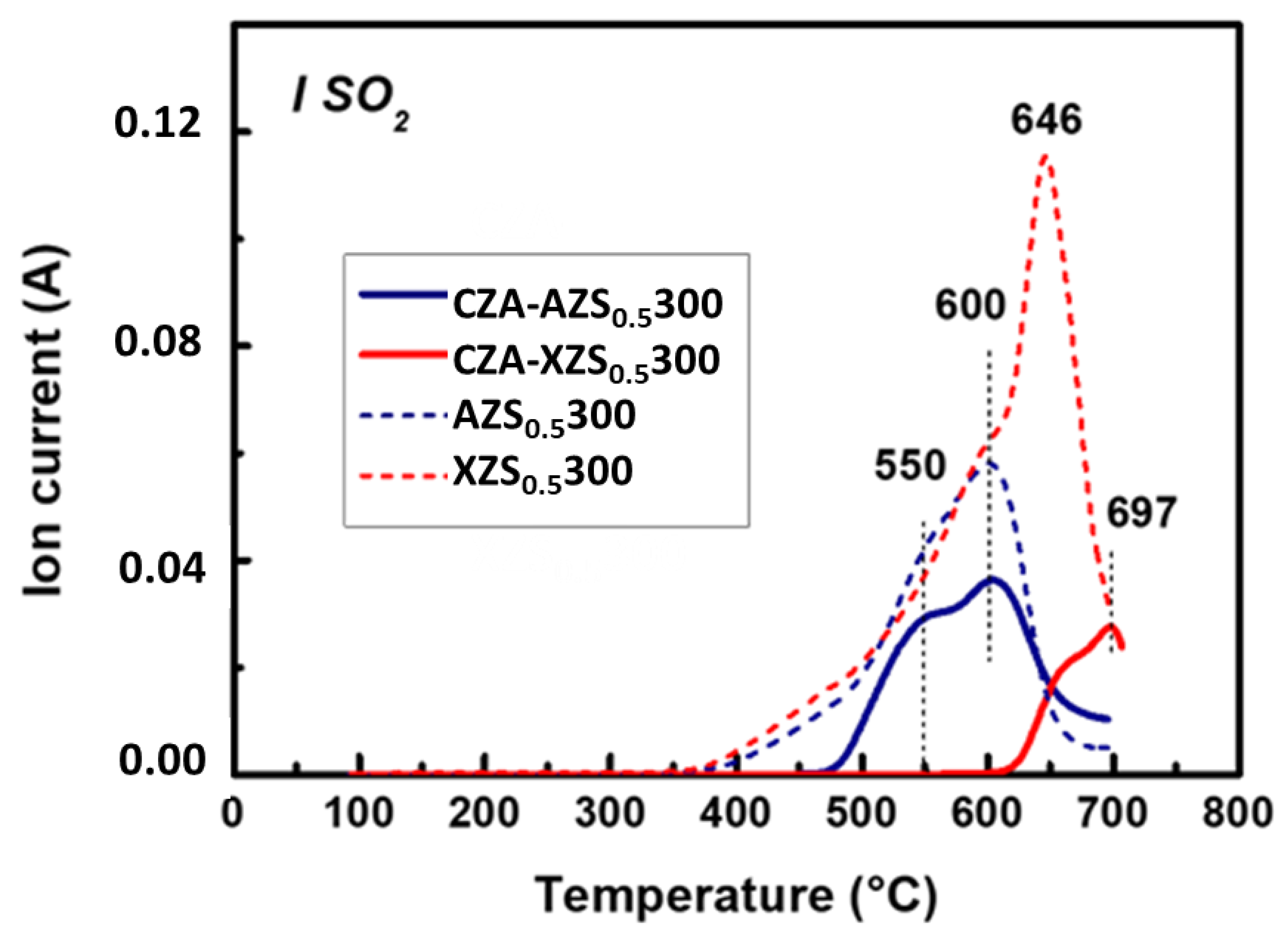
3.2.3. TPD/MS of Adsorbed NH3
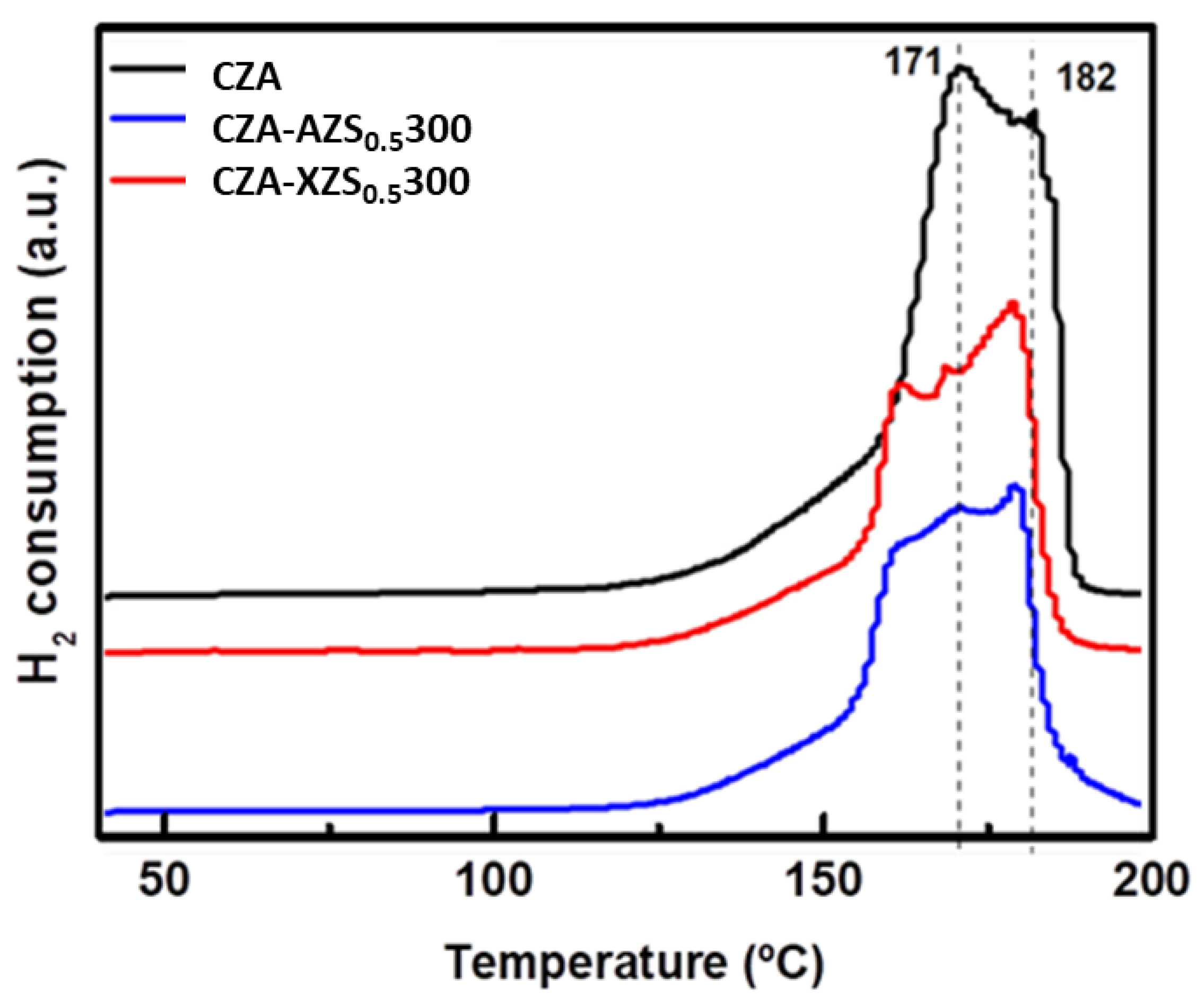
3.2.4. Temperature-Programmed Reduction (TPR-H2)
3.2.5. Structure of Reduced CZA-ZrO2/SO4 Bifunctional Composite Catalysts
3.3. Catalytic Activity in the Direct Synthesis of DME from Syngas
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thomas, G.; Feng, B.; Veeraragavan, A.; Cleary, M.J.; Drinnan, N. Emissions from DME combustion in diesel engines and their implications on meeting future emission norms: A review. Fuel Process. Technol. 2014, 119, 286–304. [Google Scholar] [CrossRef]
- Rafiee, A. Staging of di-methyl-ether (DME) synthesis reactor from synthesis gas (syngas): Direct versus indirect route. Chem. Eng. Res. Des. 2020, 163, 157–168. [Google Scholar] [CrossRef]
- Makoś, P.; Słupek, E.; Sobczak, J.; Zabrocki, D.; Hupka, J.; Rogala, A. Dimethyl ether (DME) as potential environmental friendly fuel. E3S Web Conf. 2019, 116, 48. [Google Scholar] [CrossRef]
- Fujitani, T.; Nakamura, J. The effect of ZnO in methanol synthesis catalysts on Cu dispersion and the specific activity. Catal. Lett. 1998, 56, 119–124. [Google Scholar] [CrossRef]
- Li, C.; Yuan, X.; Fujimoto, K. Development of highly stable catalyst for methanol synthesis from carbon dioxide. Appl. Catal. A Gen. 2014, 469, 306–311. [Google Scholar] [CrossRef]
- Budiman, A.; Ridwan, M.; Kim, S.M.; Choi, J.W.; Yoon, C.W.; Ha, J.M.; Suh, D.J.; Suh, Y.W. Design and preparation of high-surface-area Cu/ZnO/Al2O3 catalysts using a modified co-precipitation method for the water-gas shift reaction. Appl. Catal. A Gen. 2013, 462–463, 220–226. [Google Scholar] [CrossRef]
- Qi, G.-X.; Zheng, X.-M.; Fei, J.-H.; Hou, Z.-Y. A novel catalyst for DME synthesis from CO hydrogenation: 1. Activity, structure and surface properties. J. Mol. Catal. A Chem. 2001, 178, 195–203. [Google Scholar] [CrossRef]
- Guil-López, R.; Mota, N.; Llorente, J.; Millán, E.; Pawelec, B.; Fierro, J.L.G.; Navarro, R.M. Methanol Synthesis from CO2: A Review of the Latest Developments in Heterogeneous Catalysis. Materials 2019, 12, 3902. [Google Scholar] [CrossRef]
- Zhang, F.; Liu, Y.; Xu, X.; Yang, P.; Miao, P.; Zhang, Y.; Sun, Q. Effect of Al-containing precursors on Cu/ZnO/Al2O3 catalyst for methanol production. Fuel Process. Technol. 2018, 178, 148–155. [Google Scholar] [CrossRef]
- Vishwanathan, V.; Jun, K.W.; Kim, J.W.; Roh, H.S. Vapour phase dehydration of crude methanol to dimethyl ether over Na-modified H-ZSM-5 catalysts. Appl. Catal. A Gen. 2004, 276, 251–255. [Google Scholar] [CrossRef]
- Akarmazyan, S.S.; Panagiotopoulou, P.; Kambolis, A.; Papadopoulou, C.; Kondarides, D.I. Methanol dehydration to dimethylether over Al2O3 catalysts. Appl. Catal. B Environ. 2014, 145, 136–148. [Google Scholar] [CrossRef]
- Ramos, F.S.; de Farias, A.M.D.; Borges, L.E.P.; Monteiro, J.L.; Fraga, M.A.; Sousa-Aguiar, E.F.; Appel, L.G. Role of dehydration catalyst acid properties on one-step DME synthesis over physical mixtures. Catal. Today 2005, 101, 39–44. [Google Scholar] [CrossRef]
- Sohn, J.R. Recent advances in solid superacids. J. Ind. Eng. Chem. 2014, 10, 1–15. [Google Scholar]
- Raissi, S.; Kamoun, N.; Younes, M.K.; Ghorbel, A. Effect of drying conditions on the textural, structural and catalytic properties of Cr/ZrO2–SO4: N-hexane conversion. React. Kinet. Mech. Catal. 2015, 115, 499–512. [Google Scholar] [CrossRef]
- Witoon, T.; Permsirivanich, T.; Kanjanasoontorn, N.; Akkaraphataworn, C.; Seubsai, A.; Faungnawakij, K.; Warakulwit, C.; Chareonpanich, M.; Limtrakul, J. Direct synthesis of dimethyl ether from CO2 hydrogenation over Cu-ZnO-ZrO2/SO42−–ZrO2 hybrid catalysts: Effects of sulfur-to-zirconia ratios. Catal. Sci. Technol. 2015, 5, 2347–2357. [Google Scholar] [CrossRef]
- Reddy, B.M.; Patil, M.K. Organic syntheses and transformations catalyzed by sulfated zirconia. Chem. Rev. 2009, 109, 2185–2208. [Google Scholar] [CrossRef] [PubMed]
- Hino, M.; Kobayashi, S.; Arata, K. Solid catalyst treated with anion. 2. Reactions of butane and isobutane catalyzed by zirconium oxide treated with sulfate ion. Solid superacid catalyst. J. Am. Chem. Soc. 1979, 101, 6439–6441. [Google Scholar] [CrossRef]
- Garcia, C.; Teixeira, S.; Marciniuk, L.; Schuchardt, U. Transesterification of soybean oil catalyzed by sulfated zirconia. Bioresour. Technol. 2008, 99, 6608–6613. [Google Scholar] [CrossRef]
- Ben Hammouda, L.; Mejri, I.; Younes, M.K.; Ghorbel, A. ZrO2 Aerogels. In “Aerogels” Handbook; Aegerter, M., Leventis, N., Koebel, M., Eds.; Springer: New York, NY, USA, 2011; pp. 127–143. [Google Scholar]
- Mishra, M.K.; Tyagi, B.; Jasra, R.V. Synthesis and characterization of nano-crystalline sulfated zirconia by sol–gel method. J. Mol. Catal. A Chem. 2004, 223, 61–65. [Google Scholar] [CrossRef]
- Said, A.E.A.A.; El-Aal, M.A. Effect of different metal sulfate precursors on structural and catalytic performance of zirconia in dehydration of methanol to dimethyl ether. J. Fuel Chem. Technol. 2018, 46, 67–74. [Google Scholar] [CrossRef]
- Septiyaningrum, R.; Kurnia, A.A.; Trisunaryanti, W.; Wijaya, K. Synthesis of SO4/ZrO2 catalyst and its application on the conversion of ethanol to dimethyl ether. Iran. J. Catal. 2022, 12, 439–450. [Google Scholar]
- Khaodee, W.; Tangchupong, N.; Jongsomjit, B.; Laosiripojana, N.; Praserthdam, P.; Assabumrungrat, S. Isosynthesis via CO hydrogenation over SO4–ZrO2 catalysts. J. Ind. Eng. Chem. 2010, 16, 411–418. [Google Scholar] [CrossRef]
- Mejri, I.; Younes, M.K.; Ghorbel, A. Comparative study of the textural and structural properties of the aerogel and xerogel sulphated zirconia. J. Sol-Gel Sci. Technol. 2006, 40, 3–8. [Google Scholar] [CrossRef]
- Millán, E.; Mota, N.; Guil-López, R.; Pawelec, B.; García Fierro, J.L.; Navarro, R.M. Direct Synthesis of Dimethyl Ether from Syngas on Bifunctional Hybrid Catalysts Based on Supported H3PW12O40 and Cu-ZnO(Al): Effect of Heteropolyacid Loading on Hybrid Structure and Catalytic Activity. Catalysis 2020, 10, 1071. [Google Scholar] [CrossRef]
- Millán Ordóñez, E.; Mota, N.; Guil-López, R.; Garcia Pawelec, B.; Fierro, J.L.G.; Navarro Yerga, R.M. Direct Synthesis of Dimethyl Ether on Bifunctional Catalysts Based on Cu-ZnO(Al) and Supported H3PW12O40: Effect of Physical Mixing on Bifunctional Interactions and Activity. Ind. Eng. Chem. Res. 2021, 60, 18853–18869. [Google Scholar] [CrossRef]
- Schwan, M.; Schettler, J.; Badaczewski, F.M.; Heinrich, C.; Smarsly, B.M.; Milow, B. The effect of pulverization methods on the microstructure of stiff, ductile, and flexible carbon aerogels. J. Mater. Sci. 2020, 55, 5861. [Google Scholar] [CrossRef]
- Cychosz, K.A.; Guillet-Nicolas, R.; García-Martínez, J.; Thommes, M. Recent advances in the textural characterization of hierarchically structured nanoporous materials. Chem. Soc. Rev. 2017, 46, 389–414. [Google Scholar] [CrossRef]
- Mohammadi, M.; Shadizadeh, S.R.; Manshad, A.K.; Mohammadi, A.H. Experimental study of the relationship between porosity and surface area of carbonate reservoir rocks. J. Petrol. Explor. Prod. Technol. 2020, 10, 1817–1834. [Google Scholar] [CrossRef]
- Rabee, M.M.; Mekhemer, G.A.H.; Osatiashtiani, A.; Isaacs, M.A.; Lee, A.F.; Wilson, K.; Zaki, M.I. Acidity-Reactivity Relationships in Catalytic Esterification over Ammonium Sulfate-Derived Sulfated Zirconia. Catalysts 2017, 7, 204. [Google Scholar] [CrossRef]
- Srinivasan, R.; Keogh, R.A.; Milburn, D.R.; Davis, B.H. Sulfated Zirconia Catalysts: Characterization by TGA/DTA Mass Spectrometry. J. Catal. 1995, 153, 123–130. [Google Scholar] [CrossRef]
- Ksila, W.; Younes, M.K.; Ghorbel, A.; Rives, A. Characterization and catalytic reactivity of xerogel catalysts based on mesoporous zirconia doped with telluric acid prepared by sol–gel method: Mechanistic study of acetic acid esterification with benzyl alcohol. J. Sci. Technol. 2021, 99, 376–390. [Google Scholar] [CrossRef]
- Rachmat, A.; Trisunaryanti, W.; Saturno; Wijaya, K. Synthesis and characterization of sulfated zirconia mesopore and its application on lauric acid esterification. Mater. Renew. Sustain. Energy 2017, 6, 13. [Google Scholar] [CrossRef]
- Pérez, L.M.; Armendariz, H.; Toledo, J.A.; Hernandez, F.; Armendariz, H.; Garcia, A. n-Butane isomerization over SO42−/NiO/Al2O3/ZrO2 catalysts. Effect of the reaction pressure and metal loading. Catal. Lett. 2002, 83, 201–207. [Google Scholar] [CrossRef]
- Shi, G.; Yu, F.; Wang, Y.; Pan, D.; Wang, H.; Li, R. A novel one-pot synthesis of tetragonal sulfated zirconia catalyst with high activity for biodiesel production from the transesterification of soybean oil. Renew. Energy 2016, 92, 22–29. [Google Scholar] [CrossRef]
- Eko, A.; Brown, R. The effect of calcination temperature of sulfated zirconia catalyst for simultaneous reactions in biodiesel production. Res. J. Chem. Environ. 2018, 22, 157–162. [Google Scholar]
- Heshmatpour, F.; Aghakhanpour, R.B. Synthesis and characterization of superfine pure tetragonal nanocrystalline sulfated zirconia powder by a non-alkoxide sol–gel route. Adv. Powder. Technol. 2012, 23, 80–87. [Google Scholar] [CrossRef]
- Yu, J.; Kiwi, J.; Wang, T.; Pulgarin, C.; Rtimi, S. Evidence for a dual mechanism in the TiO2/CuxO photo-catalyst during the degradation of sulfamethazine under solar or visible light: Critical issues. J. Photochem. Photobiol. A 2019, 375, 270–279. [Google Scholar] [CrossRef]
- Prasad, K.; Pinjari, D.V.; Pandit, A.B.; Mhaske, S.T. Synthesis of zirconium dioxide by ultrasound assisted precipitation: Effect of calcination temperature. Ultrason. Sonochem. 2011, 18, 1128–1137. [Google Scholar] [CrossRef]
- Jemai, S.; Khezami, L.; Gueddana, K.; Trabelsi, K.; Hajjaji, A.; Amlouk, A.; Soucase, B.M.; Bessais, B.; Rtimi, S. Impact of Annealing on ZrO2 Nanotubes for Photocatalytic Application. Catalysts 2023, 13, 558. [Google Scholar] [CrossRef]
- Raia, R.Z.; da Silva, L.S.; Marcucci, S.M.P.; Arroyo, P.A. Biodiesel production from Jatropha curcas L. oil by simultaneous esterification and transesterification using sulphated zirconia. Catal. Today 2017, 289, 105–114. [Google Scholar] [CrossRef]
- Kumar, A.; Priyanka, M.J.; Yadav, V.; Goswami, T. Synthesis of sulfated zirconia catalyst using sol–gel technique for alkane isomerization. React. Kinet. Mech. Catal. 2022, 135, 1929–1944. [Google Scholar] [CrossRef]
- Patel, A.; Coudurier, G.; Essayem, N.; Vedrine, J.C. Effect of the addition of Sn to zirconia on the acidic properties of the sulfated mixed oxide. J. Chem. Soc. Faraday Trans. 1997, 93, 347–353. [Google Scholar] [CrossRef]
- Kim, G.; Kwon, G.; Lee, H. The role of surface hydroxyl groups on a single-atomic Rh1/ZrO2 catalyst for direct methane oxidation. Chem. Comm. 2021, 57, 1671–1674. [Google Scholar] [CrossRef]
- Deshmane, V.G.; Adewuyi, Y.G. Mesoporous nanocrystalline sulfated zirconia synthesis and its application for FFA esterification in oils. Appl. Catal. A Gen. 2013, 462–463, 196–206. [Google Scholar] [CrossRef]
- Saravanan, K.; Tyagi, B.; Bajaj, H.C. Sulfated zirconia: An efficient solid acid catalyst for esterification of myristic acid with short chain alcohols. Catal. Sci. Technol. 2012, 2, 2512–2520. [Google Scholar] [CrossRef]
- Luo, Y.; Mei, Z.; Liu, N.; Wang, H.; Han, C.; He, S. Synthesis of mesoporous sulfated zirconia nanoparticles with high surface area and their applies for biodiesel production as effective catalysts. Catal. Today 2017, 298, 99–108. [Google Scholar] [CrossRef]
- Mejri, I.; Younes, M.K.; Ghorbel, A.; Eloy, P.; Gaigneaux, E.M. Characterization and reactivity of aerogel sulfated zirconia-ceria catalyst for n-hexane isomerization. J. Porous Mater. 2009, 17, 545–551. [Google Scholar] [CrossRef]
- Katada, N. Analysis and interpretation of acidic nature of aluminosilicates. Mol. Catal. 2017, 458, 116–126. [Google Scholar] [CrossRef]
- Sekewael, S.J.; Pratika, R.A.; Hauli, L.; Amin, A.K.; Utami, M.; Wijaya, K. Recent Progress on Sulfated Nanozirconia as a Solid Acid Catalyst in the Hydrocracking Reaction. Catalysts 2022, 12, 191. [Google Scholar] [CrossRef]
- Dhachapally, N.; Sreekanth, P.; Hasyagar, U.; Nair, V.S.; Hegde, S.; Al-Mutairi, S. Metal-promoted sulfated zirconia catalysts redox and acidic characteristics and their impact on n-butane isomerization. React. Kinet. Mech. Catal. 2023, 136, 1327–1355. [Google Scholar] [CrossRef]
- Sikabwe, E.C.; Coelho, M.A.; Resasco, D.E.; White, R.L. Catalyst decomposition during temperature programmed desorption of bases from promoted sulfated zirconias. Catal. Lett. 1995, 34, 23–30. [Google Scholar] [CrossRef]
- Tyagi, B.; Mishra, M.K.; Jasra, R.V. Solvent free synthesis of 7-isopropyl-1,1-dimethyltetralin by the rearrangement of longifolene using nano-crystalline sulfated zirconia catalyst. J. Mol. Catal. A. Chem. 2009, 301, 67–78. [Google Scholar] [CrossRef]
- Lee, J. Interaction of pyridine and ammonia with a sulfate-promoted iron oxide catalyst. J. Catal. 1989, 120, 46–54. [Google Scholar] [CrossRef]
- Abu-Dahrieh, J.; Rooney, D.; Goguet, A.; Saih, Y. Activity and deactivation studies for direct dimethyl ether synthesis using CuO-ZnO-Al2O3 with NH4ZSM-5, HZSM-5 or γ-Al2O3. J. Chem. Eng. 2012, 203, 201–211. [Google Scholar] [CrossRef]
- Dasireddy, V.D.B.C.; Likozar, B. The role of copper oxidation state in Cu/ZnO/Al2O3 catalysts in CO2 hydrogenation and methanol productivity. Renew. Energy 2019, 140, 452–460. [Google Scholar] [CrossRef]
- Song, F.; Tan, Y.; Xie, H.; Zhang, Q.; Han, Y. Direct synthesis of dimethyl ether from biomass-derived syngas over Cu–ZnO–Al2O3–ZrO2(x)/γ-Al2O3 bifunctional catalysts: Effect of Zr-loading. Process. Technol. Fuel 2014, 126, 88–94. [Google Scholar] [CrossRef]
- Zhang, Y.; Fei, J.; Yu, Y.; Zheng, X. Methanol synthesis from CO2 hydrogenation over Cu based catalyst supported on zirconia modified γ- Al2O3. Energy Convers. Manag. 2006, 47, 3360–3367. [Google Scholar] [CrossRef]
- Guil-López, R.; Mota, N.; Llorente, J.; Millan, E.; Pawelec, B.G.; Fierro, J.L.G.; Navarro, R.M. Unravelling the structural modification (meso-nano-) of Cu/ZnO-Al2O3 catalysts for methanol synthesis by the residual NaNO3 in hydroxycarbonate precursors. Catalysts 2020, 10, 1346. [Google Scholar] [CrossRef]
- Melián-Cabrera, I.; López Granados, M.; Fierro, J.L.G. Structural reversibility of a ternary CuO-ZnO-Al2O3 ex hydrotalcite-containing material during wet Pd impregnation. Catal. Letters 2002, 84, 153–161. [Google Scholar] [CrossRef]
- Naik, S.P.; Du, H.; Wan, H.; Bui, V.; Miller, J.D.; Zmierczak, W.W. A Comparative Study of ZnO-CuO-Al2O3/SiO2-Al2O3 Composite and Hybrid Catalysts for Direct Synthesis of Dimethyl Ether from Syngas. Ind. Eng. Chem. Res. 2008, 47, 9791–9794. [Google Scholar] [CrossRef]
- Sai Prasad, P.S.; Wook Bae, J.; Kang, S.-H.; Lee, Y.-J.; Jun, K.-W. Single-step synthesis of DME from syngas on Cu-ZnO-Al2O3/zeolite bifunctional catalysts: The superiority of ferrierite over the other zeolites. Fuel Proc. Technol. 2008, 89, 1281–1286. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, M.J.; Kim, S.J.; Joo, O.S.; Jung, K.D. DME synthesis from synthesis gas on the admixed catalysts of Cu/ZnO/Al2O3 and ZSM-5. Appl. Catal. A Gen. 2004, 264, 37–41. [Google Scholar] [CrossRef]
- Jung, J.W.; Lee, Y.J.; Um, S.H.; Yoo, P.J.; Lee, D.H.; Jun, K.W.; Bae, J.W. Effect of copper surface area and acidic sites to intrinsic catalytic activity for dimethyl ether synthesis from biomass-derived syngas. Appl. Catal. B Environ. 2012, 126, 1–8. [Google Scholar] [CrossRef]
- Ahmed, A.I.; El-Hakam, S.A.; Samra, S.E.; EL-Khouly, A.A.; Khder, A.S. Structural characterization of sulfated zirconia and their catalytic activity in dehydration of ethanol. Colloids Surf. A Physicochem. Eng. 2008, 317, 62–70. [Google Scholar] [CrossRef]
- Rownaghi, A.A.; Rezaei, F.; Stante, M.; Hedlund, J. Selective dehydration of methanol to dimethyl ether on ZSM-5 nanocrystals. Appl. Catal. B Environ. 2012, 119–120, 56–61. [Google Scholar] [CrossRef]
- García-Trenco, A.; Martínez, A. The influence of zeolite surface-aluminum species on the deactivation of CuZnAl/zeolite hybrid catalysts for the direct DME synthesis. Catal. Today 2014, 227, 144–153. [Google Scholar] [CrossRef]
- Song, X.; Sayari, A. Sulfated Zirconia-Based Strong Solid-Acid Catalysts: Recent Progress. Catal. Rev. 1996, 38, 329–412. [Google Scholar] [CrossRef]
- Saravanan, K.; Tyagi, B.; Bajaj, H.C. Nano-crystalline, mesoporous aerogel sulfated zirconia as an efficient catalyst for esterification of stearic acid with methanol. Appl. Catal. B Environ. 2016, 192, 161–170. [Google Scholar] [CrossRef]
- Ordomsky, V.V.; Cai, M.; Sushkevich, V.; Moldovan, S.; Ersen, O.; Lancelot, C.; Valtchev, V.; Khodakov, A.Y. The role of external acid sites of ZSM-5 in deactivation of hybrid CuZnAl/ZSM-5 catalyst for direct dimethyl ether synthesis from syngas. Appl. Catal. A-Gen. 2014, 486, 266–275. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | N2 Adsorption–Desorption Isotherms a | Weight Loss b (%) | |||||
|---|---|---|---|---|---|---|---|
| SBET c (m2·g−1) | Vtotal d (m3·g−1) | Vmicro e (m3·g−1) | Smicre c (m2·g−1) | dpore (nm) | Total | SOx | |
| AZS0.5 | 201 | 0.601 | 0.001 | 4.4 | 12.0 | 58.7 | 28.6 |
| AZS0.5300 | 126 | 0.372 | 0.005 | 10.7 | 11.9 | 34.3 | 10.7 |
| AZS0.5560 | 103 | 0.355 | 0.003 | 8.2 | 13.8 | 22.4 | 12.8 |
| XZS0.5 | 169 | 0.203 | 0.023 | 46.3 | 4.8 | 19.4 | 12.2 |
| XZS0.5300 | 116 | 0.136 | 0.017 | 33.1 | 4.7 | 25.8 | 11.3 |
| XZS0.5560 | 52 | 0.083 | 0.002 | 4.7 | 6.4 | 50.8 | 17.8 |
| Catalysts | Total Acidity | Amount of Acid Sites (area/gcat) | ||
|---|---|---|---|---|
| Weak 200 < T | Moderate 250 < T < 400 | Strong >400 °C | ||
| AZS0.5 | 7.4 | 2.2 | 5.1 | 0.1 |
| AZS0.5300 | 6.8 | 1.1 | 5.6 | 0.1 |
| AZS0.5560 | 5.1 | 0.7 | 1.8 | 2.6 |
| XZS0.5 | 4.2 | 1.1 | 2.6 | 0.5 |
| XZS0.5300 | 3.5 | 1.1 | 1.8 | 0.6 |
| XZS0.5560 | 0.9 | 0.3 | 0.3 | 0.3 |
| Catalysts | Total Acidity | Amount of Acid Sites (area/gcat) | ||
|---|---|---|---|---|
| Weak 200 < T | Moderate 250 < T < 400 | Strong >400 °C | ||
| CZA-AZS0.5300 | 5.8 | 0.9 | 4.8 | 0.1 |
| AZS0.5300 | 6.8 | 1.1 | 5.6 | 0.1 |
| CZA-XZS0.5300 | 3.5 | 0.6 | 1.6 | 1.3 |
| XZS0.5300 | 3.5 | 1.1 | 1.8 | 0.6 |
| Crystallite Size (nm) | Cu0 Surface Area (m2/gcat) | |||
|---|---|---|---|---|
| CuO(Cu) | ZnO | |||
| CZA | calcined | 5.4 | 6.0 | 45.1 |
| reduced | 6.3 (6.5) a | 8.0 | ||
| used | 7.4 | 8.2 | ||
| CZA-AZS0.5300 | calcined | 4.9 | 5.4 | 33.5 |
| reduced | 6.2 (8.7) a | 6.7 | ||
| used | 6.4 | 8.1 | ||
| CZA-XZS0.5300 | calcined | 4.8 | 4.2 | 36.9 |
| reduced | 6.6 (7.9) a | 8.8 | ||
| used | 6.6 | 5.7 | ||
| Catalyst | CO Conversion (%) | Selectivity (%) | STY DME | STY CH3OH | ||
|---|---|---|---|---|---|---|
| CH3OH | DME | CO2 | ||||
| CZA | 20.9 | 100 | - | - | - | 1102 |
| CZA-AZS0.5300 | 29.8 | 8.3 | 50.9 | 40.9 | 364 | 82 |
| CZA-XZS0.5300 | 26.8 | 82.5 | 9.4 | 8.1 | 52 | 980.4 |
| Catalyst | T (°C)/P (bar) | CO Conv. (%) | Selectivity (%) | Yield DME (%) | Ref. | ||
|---|---|---|---|---|---|---|---|
| CH3OH | DME | CO2 | |||||
| CZA-AZS0.5300 | 250/30 a | 29.8 | 8.3 | 50.9 | 40.9 | 15.2 | This work |
| CZA-HZSM-5 | 250/30 a | 11.4 | 6.2 | 55.7 | 38.1 | 6.3 | [25] |
| CZA-2.7HPW/Ti | 250/30 a | 11.0 | 6.4 | 53 | 40.6 | 5.8 | [25] |
| CZA-FER | 250/40 b | 30.2 | 42.8 | 28.7 | 27.8 | 8.7 | [62] |
| CZA-NaY | 250/40 b | 14.6 | 71.7 | 12.5 | 15.2 | 1.8 | [62] |
| CZA-HY | 250/40 b | 22.7 | 12.5 | 29.7 | 57.2 | 6.7 | [62] |
| CZA-ZSM-5 | 250/40 b | 13.9 | 64.1 | 14.4 | 20.9 | 2.0 | [62] |
| CZA-FER | 250/40 b | 30.2 | 42.8 | 28.7 | 27.8 | 8.7 | [62] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lassoued, H.; Mota, N.; Millán Ordóñez, E.; Raissi, S.; Younes, M.K.; Quilis Romero, C.; Navarro Yerga, R.M. Improved Dimethyl Ether Production from Syngas over Aerogel Sulfated Zirconia and Cu-ZnO(Al) Bifunctional Composite Catalysts. Materials 2023, 16, 7328. https://doi.org/10.3390/ma16237328
Lassoued H, Mota N, Millán Ordóñez E, Raissi S, Younes MK, Quilis Romero C, Navarro Yerga RM. Improved Dimethyl Ether Production from Syngas over Aerogel Sulfated Zirconia and Cu-ZnO(Al) Bifunctional Composite Catalysts. Materials. 2023; 16(23):7328. https://doi.org/10.3390/ma16237328
Chicago/Turabian StyleLassoued, Hela, Noelia Mota, Elena Millán Ordóñez, Sahar Raissi, Mohamed Kadri Younes, Carlos Quilis Romero, and Rufino M. Navarro Yerga. 2023. "Improved Dimethyl Ether Production from Syngas over Aerogel Sulfated Zirconia and Cu-ZnO(Al) Bifunctional Composite Catalysts" Materials 16, no. 23: 7328. https://doi.org/10.3390/ma16237328