
Molecular Dynamics Simulation of the Cu3Sn/Cu Interfacial Diffusion Mechanism under Electrothermal Coupling
Abstract
:1. Introduction
2. Calculation Principle
2.1. Potential Energy Model
2.2. Verlet Algorithm
2.3. Mean Square Displacement (MSD)
2.4. Simulation Process
3. Results and Analysis
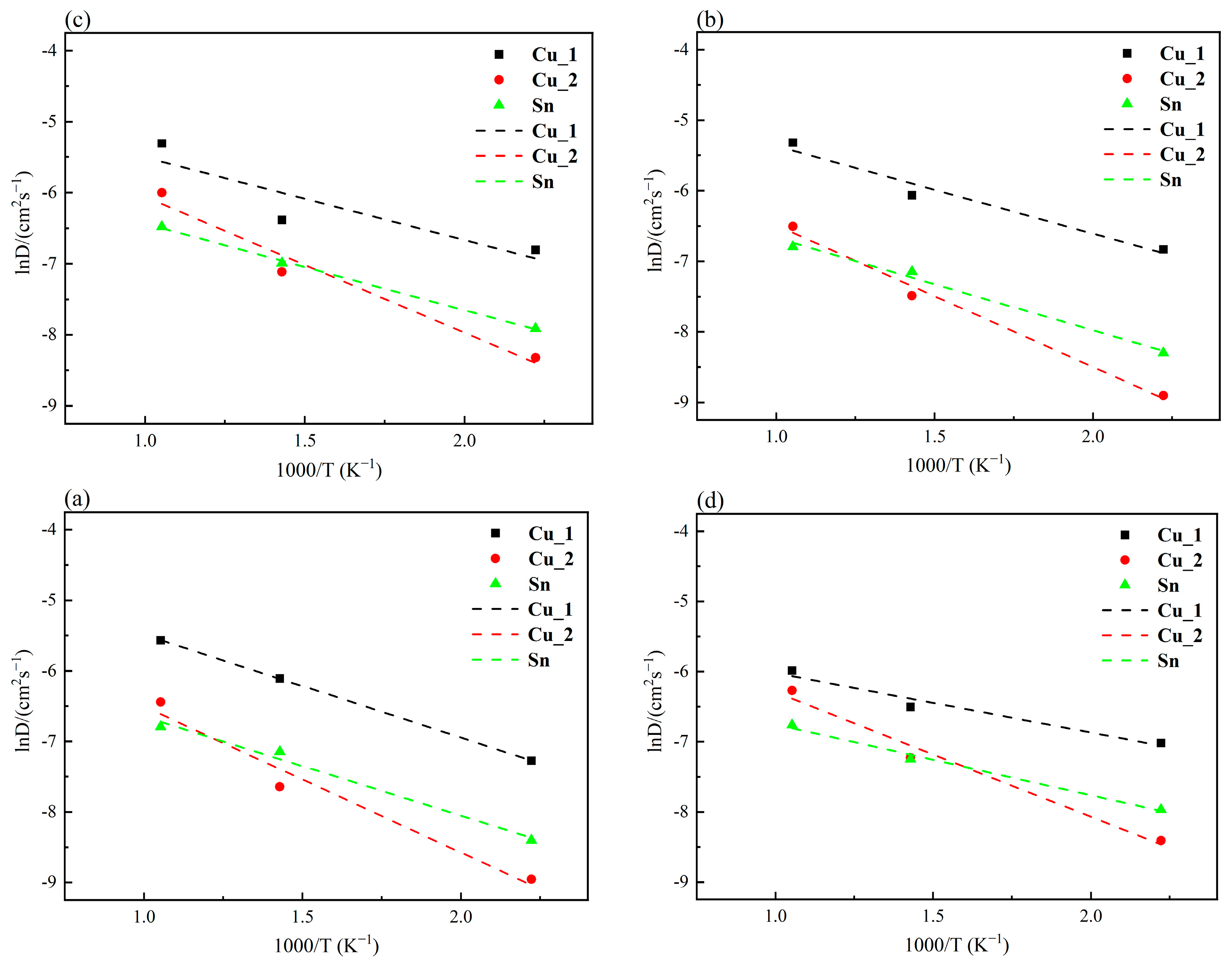
3.1. Effect of Electric Field on the Activation Energy of the Cu3Sn/Cu Interface
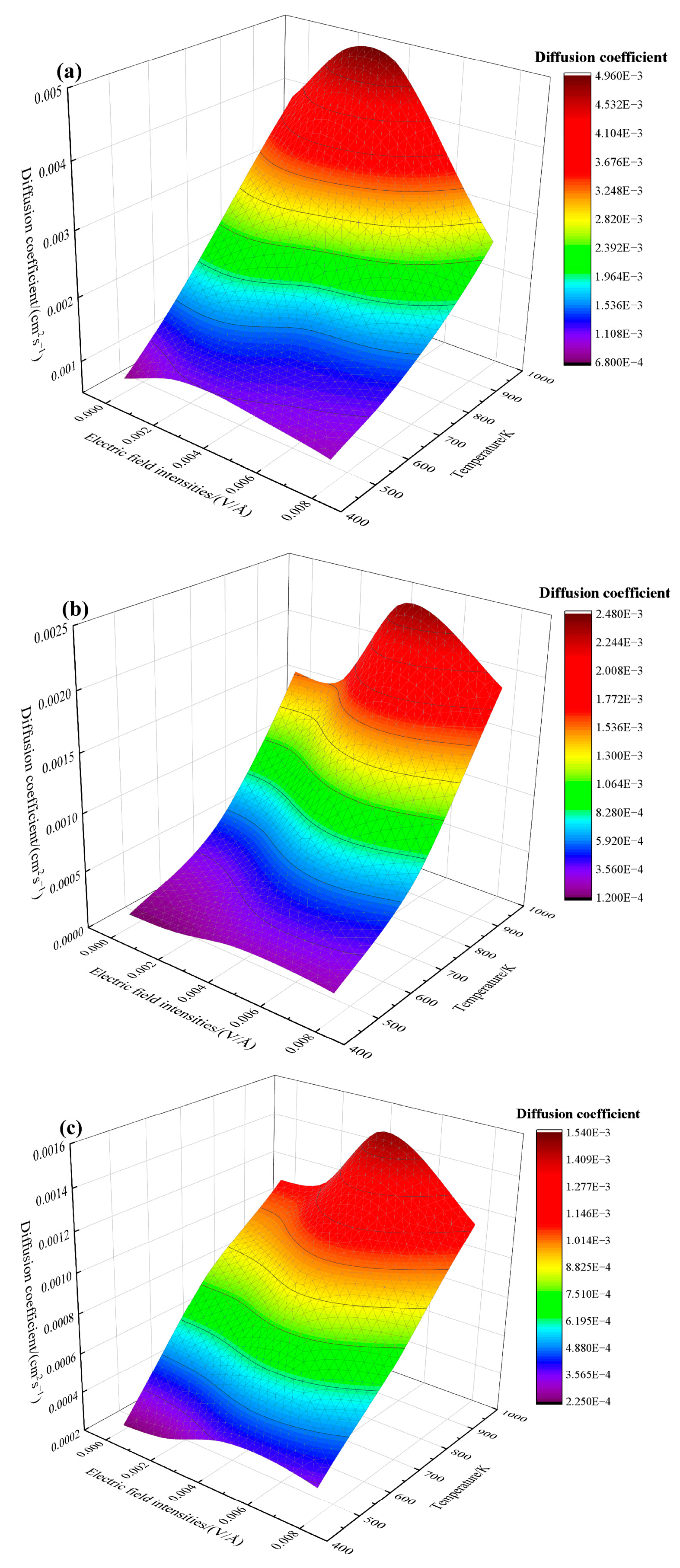
3.2. Effects of Electrothermal Coupling on the Diffusion Coefficient and Mean Square Displacement
3.3. Key Impact Factors in the Cu3Sn/Cu Interface Diffusion System under Electrothermal Coupling
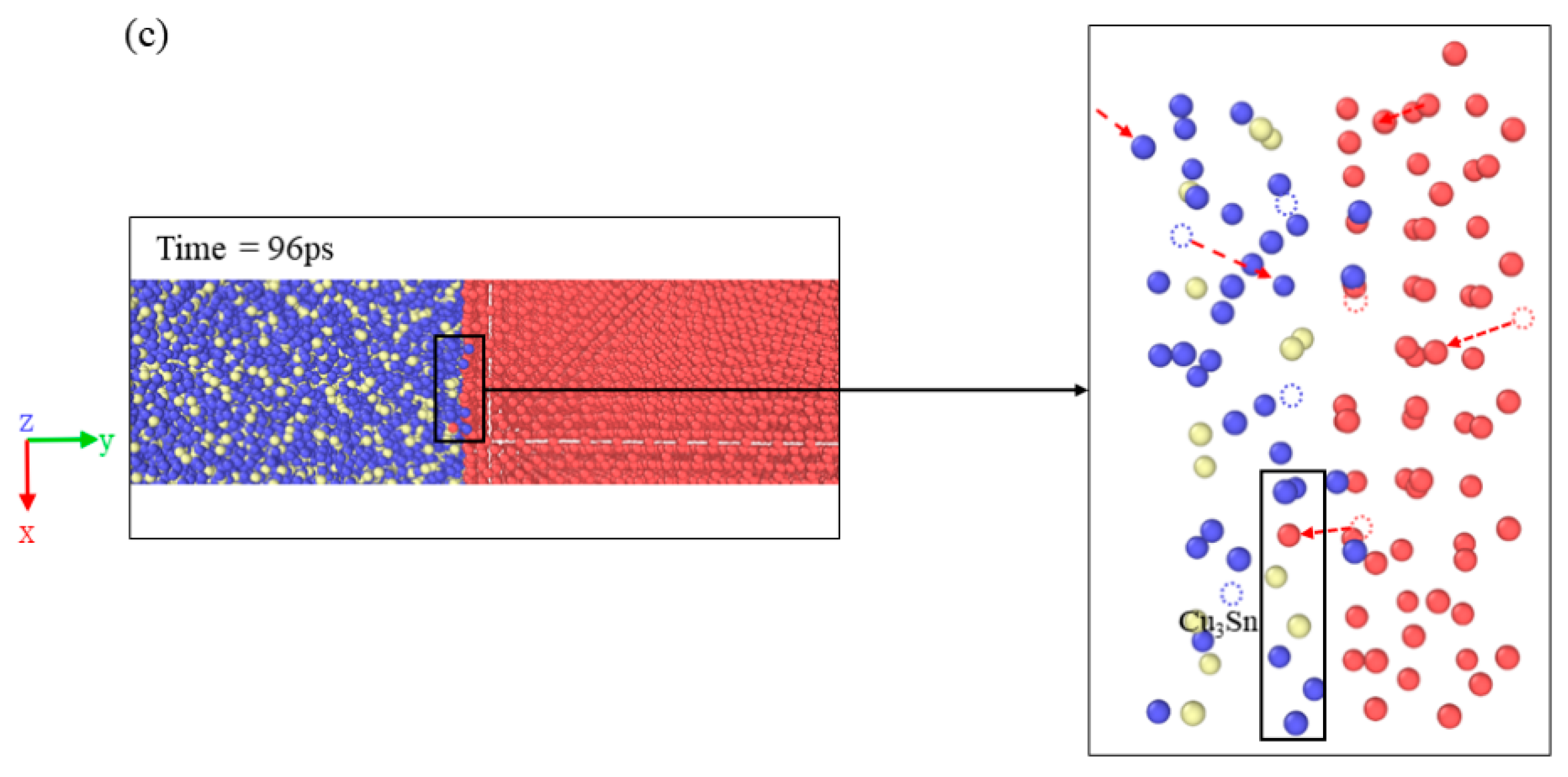
3.4. Diffusion Behavior of the Cu3Sn/Cu Interface System under Electrothermal Coupling
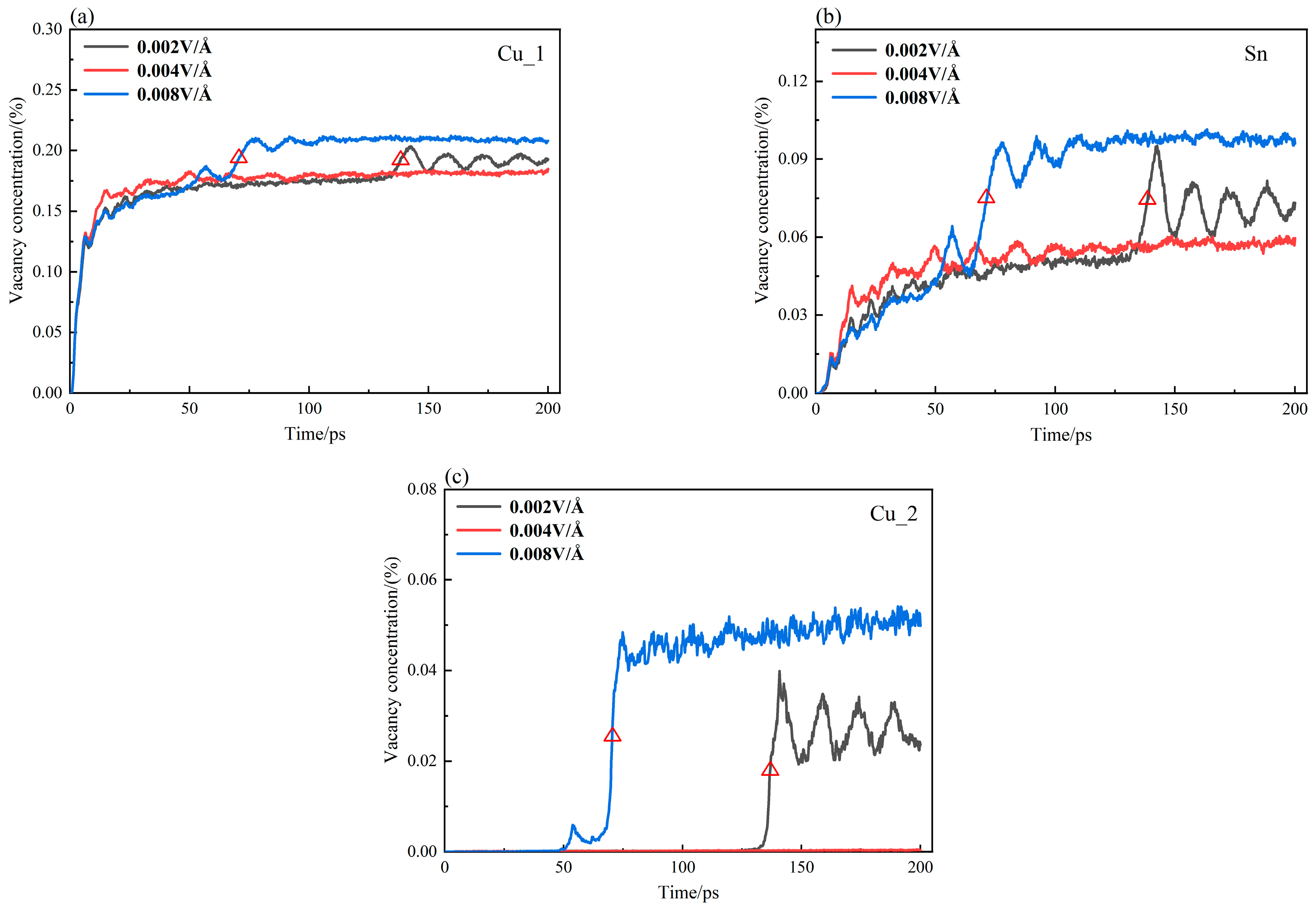
3.5. Vacancy Response Mechanism of Cu3Sn/Cu Interface Diffusion under Electrothermal Coupling
4. Discussion
5. Conclusions
- The diffusion activation energy decreases with the increase in electric field intensities, accelerating the diffusion of the Cu3Sn/Cu interface. The diffusion activation energy of the Cu_2 atoms in the Cu pad is greater than that of the Cu_1 and Sn atoms in Cu3Sn, indicating that Cu_2 atoms must overcome a more significant diffusion energy barrier to migrate.
- In the diffusion system of the Cu3Sn/Cu interface under the action of electrothermal coupling, the influence of the concentration gradient cannot be ignored. Temperature, electric field, and concentration gradient dominate the diffusion behavior.
- The contribution of electric field intensity to the diffusion coefficient increases significantly when the temperature is greater than 800 K. The increase in temperature has a more substantial effect on the diffusion enhancement of Cu_1 and Sn atoms than that of Cu_2 atoms. The diffusion of Cu_1 and Sn atoms around the interface may hinder the diffusion of Cu_2 atoms.
- The vacancy response mechanism of the Cu3Sn/Cu interface under electrothermal coupling was proposed. The electric field has direct effects on the atoms. The migration of vacancies is accelerated by the increased vacancy content as more atoms leave the equilibrium position. The lag in the abrupt variation of the vacancy concentration behind that of MSD reflects the response of vacancies to the electric field.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Nomenclature
| the structural parameter | |
| the maximum values of screening parameters | |
| the minimum values of screening parameters | |
| D | the diffusion coefficient |
| E | the electric field intensity |
| the binding energy of atom i | |
| cohesive energy of the reference structure for the I-J mixture, eV | |
| the total energy of an atomic system | |
| the formation energy of a vacancy | |
| err | |
| the embedding energy of the ith atom | |
| the electron density of atom j | |
| IMC | intermetallic compounds |
| the parameter selects the form of the function G(Gamma) used to compute the electron density | |
| MD | molecular dynamics |
| MEAM | the modified embedded atom method |
| MSD | mean square displacement |
| NVT | canonical ensemble |
| Q | the diffusion activation energy |
| R | the Boltzmann constant, JK−1 |
| the distance between atom i and atom j | |
| the equilibrium nearest-neighbor distance | |
| the cutoff radius | |
| the initial position of the atom at the initial time | |
| the position of atom i at time t | |
| the position of the atom at time t | |
| T | temperature, K |
| the start points of the abrupt change in vacancy concentration | |
| the start points of the abrupt change in MSD | |
| the endpoints of the abrupt change in vacancy concentration | |
| the endpoints of the abrupt change in MSD | |
| time span of the abrupt change in vacancy concentration | |
| time span of the abrupt change in MSD | |
| the velocity of the atom at time t | |
| the two-body interaction potential between atom i and atom j | |
| the constants related to atom j | |
| the constants related to atom j | |
| the constants related to atom k | |
| the constants related to atom k | |
| the angle between atom i, j, and k when atom i is at the center | |
| the general equation of the two-body potential | |
| the exponential decay factor for the universal energy function | |
| the exponential decay factors for the atomic densities | |
| the weighting factors for the atomic densities | |
| the electron density | |
| the concentration gradient | |
| the volume occupied by a vacancy | |
| a fraction of an atomic volume | |
| the time step | |
| v | vacancy |
| a | atom |
| vc | vacancy concentration |
| msd | mean square displacement |
References
- Cui, Z.; Fan, X.; Zhang, G. General coupling model for electromigration and one-dimensional numerical solutions. J. Appl. Phys. 2019, 125, 105101. [Google Scholar] [CrossRef]
- Fan, D.; Liu, Z.; Huang, Y.; Wang, X. Structure and properties of arc assisted activating TIG welding. Trans. China Weld. Inst. 2014, 35, 1–5. (In Chinese) [Google Scholar]
- Min, S. Study on the Growth and Control Mechanism of Cu3Sn in the Sn/Cu Soldering Interface. Master’s Thesis, Dalian University of Technology, Dalian, China, 2022. (In Chinese). [Google Scholar]
- Bader, S.; Gust, W.; Hieber, H. Rapid formation of intermetallic compounds interdiffusion in the Cu-Sn and Ni-Sn systems. Acta Metall. Mater. 1995, 43, 329–337. [Google Scholar]
- Lee, C.C.; Chen, Y.C. High temperature tin-copper joints produced at low process temperature for stress reduction. Thin Solid Film. 1996, 286, 213–218. [Google Scholar] [CrossRef]
- Liang, L.; Zhang, J.; Xu, Y.; Zhang, Y.; Wang, W.; Yang, J. The effect of pressure and orientation on Cu-Cu3Sn interface reliability under isothermal ageing and monotonic traction via molecular dynamics investigation. Mater. Des. 2018, 149, 194–204. [Google Scholar] [CrossRef]
- Wang, J.; Chen, J.; Zhang, L.; Zhang, Z.; Han, Y.; Hu, X.; Lu, H.; Zhang, S. Forming mechanism and growth of Kirkendall voids of Sn/Cu joints for electronic packaging: A recent review. J. Adv. Join. Process. 2022, 6, 100125. [Google Scholar] [CrossRef]
- Zhang, L.; Li, J.; Yin, L.; Zhang, H.; Xu, M.; Yao, Z. Phase Field Simulation of Intermetallic Compound Evolution at the Interface of Copper-Tin Coating Under Thermal-Mechanical-Electrical Diffusion Coupling. J. Electron. Mater. 2023, 52, 3321–3333. [Google Scholar] [CrossRef]
- Du, Y.; Gao, L.Y.; Yu, D.; Liu, Z.-Q. Comparison and mechanism of electromigration reliability between Cu wire and Au wire bonding in molding state. J. Mater. Sci. Mater. Electron. 2020, 31, 2967–2975. [Google Scholar] [CrossRef]
- Xu, K.; Fu, X.; Wang, X.; Fu, Z.; Yang, X.; Chen, S.; Shi, Y.; Huang, Y.; Chen, H. The effect of grain orientation of β-Sn on Copper pillar solder joints during electromigration. Materials 2021, 15, 108. [Google Scholar] [CrossRef]
- Liu, X.; Xiao, L.; Peng, Y.; Xu, H. Effect of Electromigration on Morphology and Shear Property of Full Cu3Sn Solder Joints. Hot Work. Technol. 2020, 49, 8–11. (In Chinese) [Google Scholar]
- Wang, L.; Cao, Z.H.; Xu, J.H.; Yu, L.H.; Huang, T.; Meng, X.K. Influence of electric field annealing on atom diffusion in Cu/Ta/Si stacks. Appl. Phys. A 2014, 114, 1091–1095. [Google Scholar] [CrossRef]
- Wang, L.; Cao, Z.H.; Hu, K.; She, Q.; Meng, X. Effects of electric field annealing on the interface diffusion of Cu/Ta/Si stacks. Appl. Surf. Sci. 2011, 257, 10845–10849. [Google Scholar] [CrossRef]
- Zhang, L.; Xiong, D.; Su, Z.; Li, J.; Yin, L.; Yao, Z.; Wang, G.; Zhang, L.; Zhang, H. Molecular dynamics simulation and experimental study of tin growth in SAC lead-free microsolder joints under thermo-mechanical-electrical coupling. Mater. Today Commun. 2022, 33, 104301. [Google Scholar] [CrossRef]
- Li, B.; Zhang, Z.; Zhou, X.; Liu, M.; Jie, Y. Mechanical behavior and microstructure evolution of Al/AlCu alloy interface. J. Mater. Sci. 2023, 58, 5489–5502. [Google Scholar] [CrossRef]
- Chen, J.; Xu, M.; Jin, Y.; Wang, K.-Y.; Chun, Y.; Lu, H. Effect of electric field and Sn grain orientation on Cu consumption in Sn/Cu solder joint. Comput. Mater. Sci. 2014, 95, 166–171. [Google Scholar] [CrossRef]
- Zhang, L.; Xiong, D.; Li, J.; Yin, L.; Yao, Z.; Wang, G.; Zhang, L.; Zhang, H. Molecular dynamics simulation of the interfacial evolution and whisker growth of copper-tin coating under electrothermal coupling. Comput. Mater. Sci. 2022, 202, 110981. [Google Scholar] [CrossRef]
- Yu, B.; Li, X.; Yao, P.; Zhu, Y. Numerical simulation of diffusion behavior of Cu3Sn/Cu interface based on molecular dynamics. Trans. China Weld. Inst. 2017, 38, 50–54. (In Chinese) [Google Scholar]
- Guo, L.; Li, X.; Yao, P.; Li, Y. Molecular Dynamics Simulation of Diffusion Behavior of Atoms at the Interface of Cu/Cu3Sn Under Electric Field. Mater. Rep. 2020, 34, 2137–2141. [Google Scholar]
- Baskes, M.I. Modified embedded-atom potentials for cubic materials and impurities. Phys. Rev. B 1992, 46, 2727–2742. [Google Scholar] [CrossRef]
- Cheng, H.C.; Yu, C.F.; Chen, W.H. Strain- and strain-rate-dependent mechanical properties and behaviors of Cu3Sn compound using molecular dynamics simulation. J. Mater. Sci. 2012, 47, 3103–3114. [Google Scholar] [CrossRef]
- Baskes, M.I.; Angelo, J.E.; Bisson, C.L. Atomistic calculations of composite interfaces. Model. Simul. Mater. Sci. Eng. 1994, 2, 505–518. [Google Scholar] [CrossRef]
- Wang, X.; Chang, J.; Gao, D.; Fang, N.; Zhang, S.; Yang, X.; Wen, G.; Wu, S.; Long, W.; He, P. Molecular Dynamics Simulation of High-Nitrogen Stainless Steel Brazed by AgCuNi Filler Metal. Rare Met. Mater. Eng. 2022, 51, 3223–3229. [Google Scholar]
- Daeumer, M.; Sandoval, E.D.; Azizi, A.; Bagheri, M.H.; Bae, I.-T.; Panta, S.; Koulakova, E.A.; Cotts, E.; Arvin, C.L.; Kolmogorov, A.N.; et al. Orientation-dependent transport properties of Cu3Sn. Acta Mater. 2022, 227, 117671. [Google Scholar] [CrossRef]
- Cao, Z.H.; Wang, F.; Wang, L.; Meng, X.-K. Coupling effect of the electric and temperature fields on the growth of nanocrystalline copper films. Phys. Rev. B 2010, 81, 113405. [Google Scholar] [CrossRef]
- Wang, L.; Cao, Z.H.; Syed, J.A.; Hu, K.; She, Q.W.; Meng, X.K. Electric field accelerating interface diffusion in Cu/Ru/TaN/Si stacks during annealing. Electrochem. Solid-State Lett. 2012, 15, H188. [Google Scholar] [CrossRef]
- Hutchinson, B. Critical Assessment 39: Effects of applied fields during annealing of metals-a review. Mater. Sci. Technol. 2020, 36, 1125–1138. [Google Scholar] [CrossRef]
- Paul, A.; Ghosh, C.; Boettinger, W.J. Diffusion Parameters and Growth Mechanism of Phases in the Cu-Sn System. Metall. Mater. Trans. A 2011, 42, 952–963. [Google Scholar] [CrossRef]
- Wang, H.; Kou, R.; Harrington, T.; Vecchio, K.S. Electromigration effect in Fe-Al diffusion couples with field-assisted sintering. Acta Mater. 2020, 186, 631–643. [Google Scholar] [CrossRef]
- Huntington, H.B.; Grone, A.R. Current-induced marker motion in gold wires. J. Phys. Chem. Solids 1961, 20, 76–87. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Wang, Y.; Fang, Y. Effect of Grain Structure and Ni/Au-UBM Layer on Electromigration-Induced Failure Mechanism in Sn-3.0 Ag-0.5 Cu Solder Joints. Micromachines 2022, 13, 953. [Google Scholar] [CrossRef]
- Chen, G.; Cui, X.; Wu, Y.; Li, W.; Wu, F. Microstructural, compositional and hardness evolutions of 96.5 Sn-3Ag-0.5 Cu/TiC composite solder under thermo-migration stressing. J. Mater. Sci. Mater. Electron. 2020, 31, 9492–9503. [Google Scholar] [CrossRef]
- Cui, Z.; Fan, X.; Zhang, G. Molecular dynamic study for concentration-dependent volume relaxation of vacancy. Microelectron. Reliab. 2021, 120, 114127. [Google Scholar] [CrossRef]
- Akbarzadeh, A.R.; Chen, Z.Z.; Kioussis, N. Crucial role of surface instability and mobility of vacancy clusters in metals. Phys. Rev. B 2009, 79, 195404. [Google Scholar] [CrossRef]
- Yuan, X.J.; Chen, N.X.; Shen, J.; Hu, W. Embedded-atom-method in-teratomic potentials from lattice inversion. J. Phys. Condens. Matter 2010, 22, 375503. [Google Scholar] [CrossRef] [PubMed]
- Ren, E.; Li, X.; Zhang, H.; Han, X. Effect of vacancy on element diffusion in Cu3Sn layer for Cu/Sn solder joints. Electron. Compon. Mater. 2022, 41, 381–386. (In Chinese) [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | A | R0 (Å) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cu | 3.62 | 1.07 | 2.50 | 5.11 | 3.62 | 2.20 | 6.00 | 2.20 | 3.14 | 2.49 | 2.95 | 1.00 | 0 |
| Sn | 3.08 | 1.00 | 3.44 | 6.20 | 6.20 | 6.00 | 6.00 | 6.00 | 4.50 | 6.50 | −0.18 | 1.00 | 0 |
| Cu3Sn | 3.50 | 2.68 | 5.38 |
| ijk | ||||||
|---|---|---|---|---|---|---|
| 111 | 112 | 212 | 121 | 221 | 222 | |
| 2.8 | 2.8 | 2.8 | 2.8 | 2.8 | 2.8 | |
| 2.0 | 0.8 | 0.8 | 0.8 | 0.8 | 0.8 | |
| Electric Field Intensity (V/Å) | Cu_1 | Cu_2 | Sn | ||||||
|---|---|---|---|---|---|---|---|---|---|
| err (%) | err (%) | err (%) | |||||||
| 0.002 | 9.514 | 9.901 | 4.07 | 9.987 | 9.557 | 4.31 | 11.925 | 11.537 | 3.25 |
| 0.008 | 11.537 | 11.925 | 3.36 | 24.548 | 25.141 | 2.42 | 13.560 | 12.700 | 6.35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Z.; Lan, X.; Li, L.; Cheng, Y. Molecular Dynamics Simulation of the Cu3Sn/Cu Interfacial Diffusion Mechanism under Electrothermal Coupling. Materials 2023, 16, 7507. https://doi.org/10.3390/ma16247507
He Z, Lan X, Li L, Cheng Y. Molecular Dynamics Simulation of the Cu3Sn/Cu Interfacial Diffusion Mechanism under Electrothermal Coupling. Materials. 2023; 16(24):7507. https://doi.org/10.3390/ma16247507
Chicago/Turabian StyleHe, Zhiwei, Xin Lan, Lezhou Li, and Yong Cheng. 2023. "Molecular Dynamics Simulation of the Cu3Sn/Cu Interfacial Diffusion Mechanism under Electrothermal Coupling" Materials 16, no. 24: 7507. https://doi.org/10.3390/ma16247507