
Reasonable Design of MXene-Supported Dual-Atom Catalysts with High Catalytic Activity for Hydrogen Evolution and Oxygen Evolution Reaction: A First-Principles Investigation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
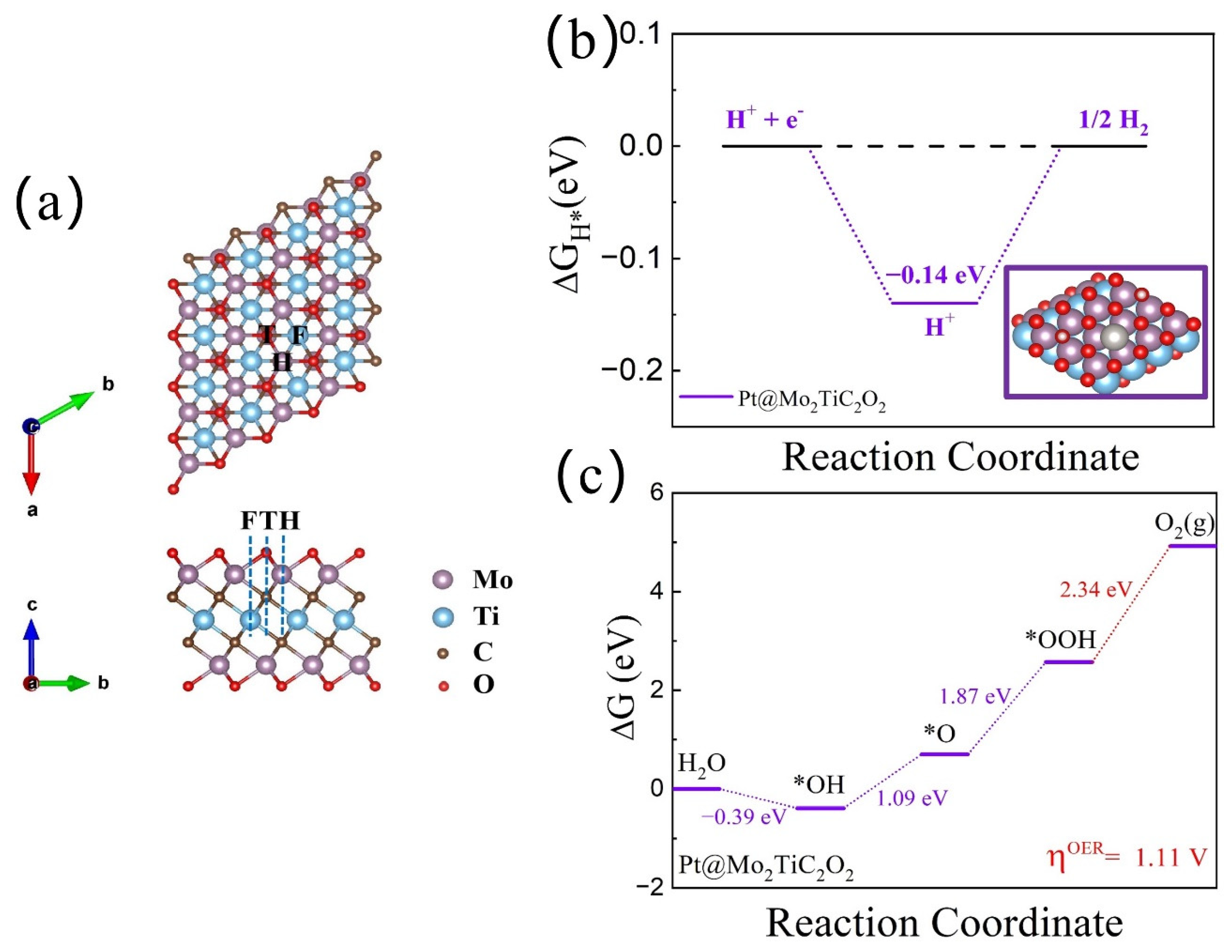
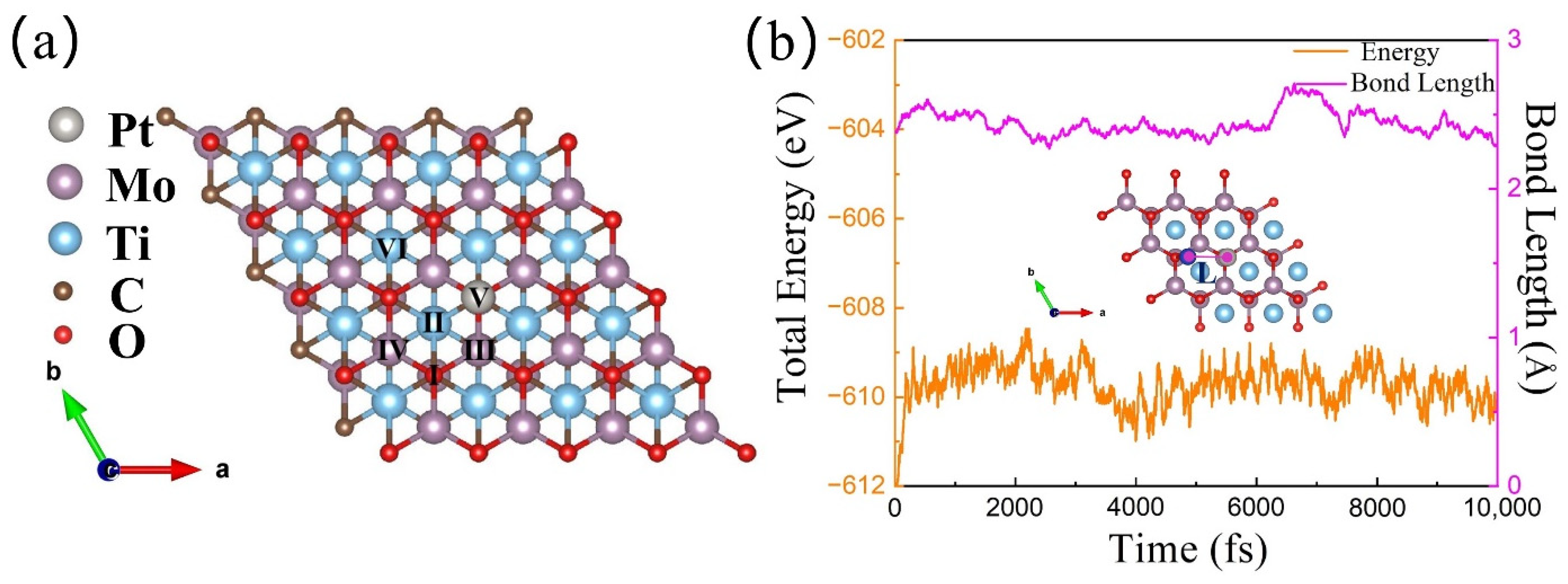
3.1. Stability of MXene-Supported Catalysts
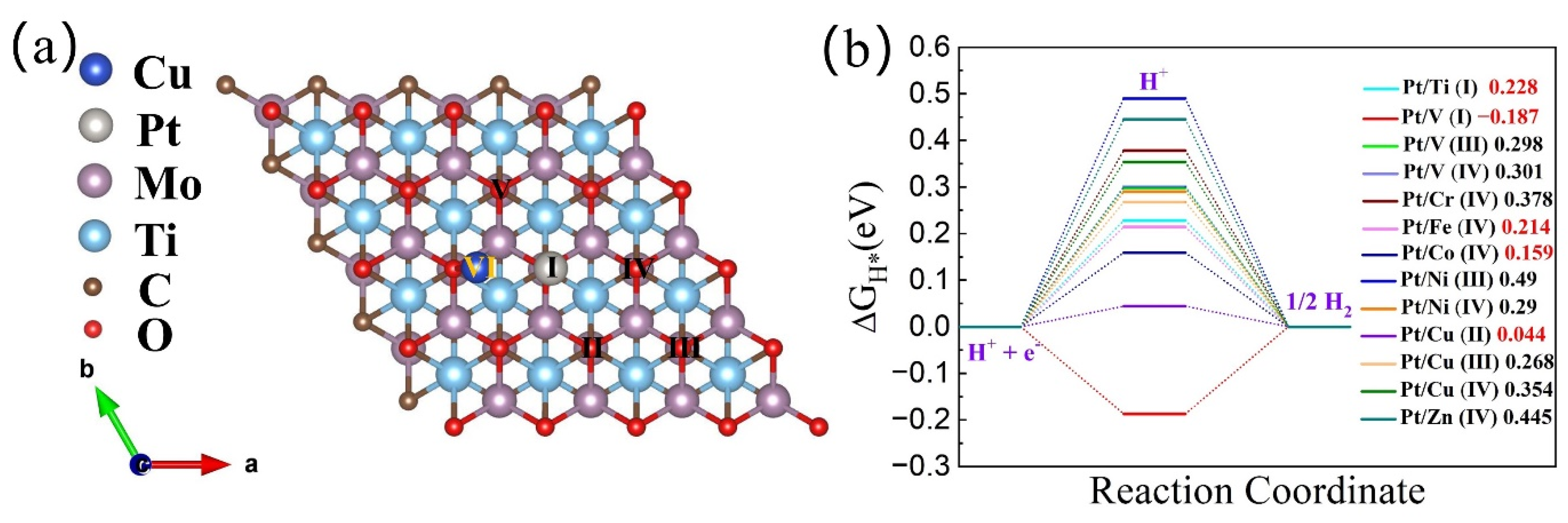
3.2. HER Catalytic Activity of h-DACs
3.3. OER Catalytic Activity of h-DACs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheng, Y.; Dai, J.; Song, Y.; Zhang, Y. Nanostructure of Cr2CO2 MXene Supported Single Metal Atom as an Efficient Bifunctional Electrocatalyst for Overall Water Splitting. ACS Appl. Energy Mater. 2019, 2, 6851–6859. [Google Scholar] [CrossRef]
- Fu, Z.; Ling, C.; Wang, J. A Ti3C2O2 Supported Single Atom, Trifunctional Catalyst for Electrochemical Reactions. J. Mater. Chem. A 2020, 8, 7801–7807. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, B.; Peng, Q.; Zhou, J.; Sun, Z. Mo2B2 MBene-Supported Single-Atom Catalysts as Bifunctional HER/OER and OER/ORR Electrocatalysts. J. Mater. Chem. A 2021, 9, 433–441. [Google Scholar] [CrossRef]
- Ling, C.; Shi, L.; Ouyang, Y.; Zeng, X.C.; Wang, J. Nanosheet Supported Single-Metal Atom Bifunctional Catalyst for Overall Water Splitting. Nano Lett. 2017, 17, 5133–5139. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, X.; Zhang, G.; Shi, P.; Wang, A.-L. Rational Catalyst Design for Oxygen Evolution under Acidic Conditions: Strategies toward Enhanced Electrocatalytic Performance. J. Mater. Chem. A 2021, 9, 5890–5914. [Google Scholar] [CrossRef]
- Ahsan, M.A.; He, T.; Eid, K.; Abdullah, A.M.; Curry, M.L.; Du, A.; Santiago, A.R.P.; Echegoyen, L.; Noveron, J.C. Tuning the Intermolecular Electron Transfer of Low-Dimensional and Metal-Free BCN/C60 Electrocatalysts via Interfacial Defects for Efficient Hydrogen and Oxygen Electrochemistry. J. Am. Chem. Soc 2021, 143, 1203–1215. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Abdelgawad, A.; Li, J.; Eid, K. Non-Metal-Doped Porous Carbon Nitride Nanostructures for Photocatalytic Green Hydrogen Production. Int. J. Mol. Sci. 2022, 23, 15129. [Google Scholar] [CrossRef]
- Ahsan, M.A.; He, T.; Eid, K.; Abdullah, A.M.; Sanad, M.F.; Aldalbahi, A.; Alvarado-Tenorio, B.; Du, A.; Santiago, A.R.P.; Noveron, J.C. Controlling the Interfacial Charge Polarization of MOF-De-rived 0D−2D vdW Architectures as a Unique Strategy for Bifunctional Oxygen Electrocatalysis. ACS Appl. Mater. Interfaces 2022, 14, 3919–3929. [Google Scholar] [CrossRef]
- Reier, T.; Oezaslan, M.; Strasser, P. Electrocatalytic Oxygen Evolution Reaction (OER) on Ru, Ir, and Pt Catalysts: A Comparative Study of Nanoparticles and Bulk Materials. ACS Catal. 2012, 2, 1765–1772. [Google Scholar] [CrossRef]
- Sanchez Casalongue, H.G.; Ng, M.L.; Kaya, S.; Friebel, D.; Ogasawara, H.; Nilsson, A. In Situ Observation of Surface Species on Iridium Oxide Nanoparticles during the Oxygen Evolution Reaction. Angew. Chem. Int. Ed. 2014, 53, 7169–7172. [Google Scholar] [CrossRef] [Green Version]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23–J26. [Google Scholar] [CrossRef]
- Naguib, M.; Kurtoglu, M.; Presser, V.; Lu, J.; Niu, J.; Heon, M.; Hultman, L.; Gogotsi, Y.; Barsoum, M.W. Two-Dimensional Nanocrystals Produced by Exfoliation of Ti3AlC2. Adv. Mater. 2011, 23, 4248–4253. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhang, W. High Throughput Screening of M3C2 MXenes for Efficient CO2 Reduction Conversion into Hydrocarbon Fuels. Nanoscale 2020, 12, 7660–7673. [Google Scholar] [CrossRef]
- Handoko, A.D.; Chen, H.; Lum, Y.; Zhang, Q.; Anasori, B.; Seh, Z.W. Two-Dimensional Titanium and Molybdenum Carbide MXenes as Electrocatalysts for CO2 Reduction. iScience 2020, 23, 101181. [Google Scholar] [CrossRef]
- Huang, B.; Li, N.; Ong, W.-J.; Zhou, N. Single Atom-Supported MXene: How Single-Atomic-Site Catalysts Tune the High Activity and Selectivity of Electrochemical Nitrogen Fixation. J. Mater. Chem. A 2019, 7, 27620–27631. [Google Scholar] [CrossRef]
- Gan, J.; Li, F.; Tang, Y.; Tang, Q. Theoretical Study of Transition-Metal-Modified Mo2CO2 MXene as a Catalyst for the Hydrogen Evolution Reaction. ChemSusChem 2020, 13, 6005–6015. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhou, J.; Sun, Z. Novel 2D Transition-Metal Carbides: Ultrahigh Performance Electrocatalysts for Overall Water Splitting and Oxygen Reduction. Adv. Funct. Mater. 2020, 30, 2000570. [Google Scholar] [CrossRef]
- Zhou, S.; Yang, X.; Pei, W.; Jiang, Z.; Zhao, J. MXene and MBene as Efficient Catalysts for Energy Conversion: Roles of Surface, Edge and Interface. J. Phys. Energy 2021, 3, 012002. [Google Scholar] [CrossRef]
- Eid, K.; Lu, Q.; Abdel-Azeim, S.; Soliman, A.; Abdullah, A.M.; Abdelgwad, A.M.; Forbes, R.P.; Ozoemena, K.I.; Varma, R.S.; Shibl, M.F. Highly exfoliated Ti3C2Tx MXene nanosheets atomically doped with Cu for efficient electrochemical CO2 reduction: An experimental and theoretical study. J. Mater. Chem. A 2022, 10, 1965–1975. [Google Scholar] [CrossRef]
- Peng, Q.; Rehman, J.; Eid, K.; Alofi, A.S.; Laref, A.; Albaqami, M.D.; Alotabi, R.G.; Shibl, M.F. Vanadium Carbide (V4C3) MXene as an Effificient Anode for Li-Ion and Na-Ion Batteries. Nanomaterials 2022, 12, 2825. [Google Scholar] [CrossRef]
- Ibrahim, Y.; Meslam, M.; Eid, K.; Salah, B.; Abdullah, A.M.; Ozoemena, K.I.; Elzatahry, A.; Sharaf, M.; Sillanpää, M. A Review of MXenes as Emergent Materials for Dye Removal from Wastewater. Sep. Purif. Technol. 2022, 282, 120083. [Google Scholar] [CrossRef]
- Luo, Y.; Chen, G.-F.; Ding, L.; Chen, X.; Ding, L.-X.; Wang, H. Efficient Electrocatalytic N2 Fixation with MXene under Ambient Conditions. Joule 2019, 3, 279–289. [Google Scholar] [CrossRef]
- Lu, C.; Yang, L.; Yan, B.; Sun, L.; Zhang, P.; Zhang, W.; Sun, Z. Nitrogen-Doped Ti3C2 MXene: Mechanism Investigation and Electrochemical Analysis. Adv. Funct. Mater. 2020, 30, 2000852. [Google Scholar] [CrossRef]
- Shahzad, F.; Iqbal, A.; Kim, H.; Koo, C.M. 2D Transition Metal Carbides (MXenes): Applications as an Electrically Conducting Material. Adv. Mater. 2020, 32, 2002159. [Google Scholar] [CrossRef]
- Gogotsi, Y.; Anasori, B. The Rise of MXenes. ACS Nano 2019, 13, 8491–8494. [Google Scholar] [CrossRef]
- Anasori, B.; Lukatskaya, M.R.; Gogotsi, Y. 2D Metal Carbides and Nitrides (MXenes) for Energy Storage. Nat. Rev. Mater. 2017, 2, 16098. [Google Scholar] [CrossRef]
- Pang, J.; Mendes, R.G.; Bachmatiuk, A.; Zhao, L.; Ta, H.Q.; Gemming, T.; Liu, H.; Liu, Z.; Rummeli, M.H. Applications of 2D MXenes in Energy Conversion and Storage Systems. Chem. Soc. Rev. 2019, 48, 72–133. [Google Scholar] [CrossRef]
- Kan, D.; Wang, D.; Zhang, X.; Lian, R.; Xu, J.; Chen, G.; Wei, Y. Rational Design of Bifunctional ORR/OER Catalysts Based on Pt/Pd-Doped Nb2CT2 MXene by First-Principles Calculations. J. Mater. Chem. A 2020, 8, 3097–3108. [Google Scholar] [CrossRef]
- Zhang, M.; Du, J.; Chen, Y. Single Cu Atom Supported on Modified H-BN Monolayer as n-p Co-doped Catalyst for CO Oxidation: A Computational Study. Catal. Today 2021, 368, 148–160. [Google Scholar] [CrossRef]
- Lei, Y.; Wang, Y.; Liu, Y.; Song, C.; Li, Q.; Wang, D.; Li, Y. Designing Atomic Active Centers for Hydrogen Evolution Electrocatalysts. Angew. Chem. Int. Ed. 2020, 59, 20794–20812. [Google Scholar] [CrossRef]
- Ouyang, Y.; Ling, C.; Chen, Q.; Wang, Z.; Shi, L.; Wang, J. Activating Inert Basal Planes of MoS2 for Hydrogen Evolution Reaction through the Formation of Different Intrinsic Defects. Chem. Mater. 2016, 28, 4390–4396. [Google Scholar] [CrossRef]
- Jung, E.; Shin, H.; Lee, B.-H.; Efremov, V.; Lee, S.; Lee, H.S.; Kim, J.; Hooch Antink, W.; Park, S.; Lee, K.-S.; et al. Atomic-Level Tuning of Co–N–C Catalyst for High-Performance Electrochemical H2O2 Production. Nat. Mater. 2020, 19, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Kour, G.; Mao, X.; Du, A. Single Copper Atoms Supported on ZnS as an Efficient Catalyst for Electrochemical Reduction of CO to CH3OH. ChemNanoMat 2020, 6, 1806–1811. [Google Scholar] [CrossRef]
- Xu, H.; Wang, D.; Yang, P.; Liu, A.; Li, R.; Li, Y.; Xiao, L.; Ren, X.; Zhang, J.; An, M. Atomically Dispersed M–N–C Catalysts for the Oxygen Reduction Reaction. J. Mater. Chem. A 2020, 8, 23187–23201. [Google Scholar] [CrossRef]
- Chen, Z.; Fan, X.; Shen, Z.; Ruan, X.; Wang, L.; Zeng, H.; Wang, J.; An, Y.; Hu, Y. Cu Anchored Ti2NO2 as High Performance Electrocatalyst for Oxygen Evolution Reaction: A Density Functional Theory Study. ChemCatChem 2020, 12, 4059–4066. [Google Scholar] [CrossRef]
- Choi, C.H.; Kim, M.; Kwon, H.C.; Cho, S.J.; Yun, S.; Kim, H.-T.; Mayrhofer, K.J.J.; Kim, H.; Choi, M. Tuning Selectivity of Electrochemical Reactions by Atomically Dispersed Platinum Catalyst. Nat. Commun. 2016, 7, 10922. [Google Scholar] [CrossRef]
- Li, J.; Chen, S.; Yang, N.; Deng, M.; Ibraheem, S.; Deng, J.; Li, J.; Li, L.; Wei, Z. Ultrahigh-Loading Zinc Single-Atom Catalyst for Highly Efficient Oxygen Reduction in Both Acidic and Alkaline Media. Angew. Chem. Int. Ed. 2019, 58, 7035–7039. [Google Scholar] [CrossRef]
- Liu, P.; Zhao, Y.; Qin, R.; Mo, S.; Chen, G.; Gu, L.; Chevrier, D.M.; Zhang, P.; Guo, Q.; Zang, D.; et al. Photochemical Route for Synthesizing Atomically Dispersed Palladium Catalysts. Science 2016, 352, 797–800. [Google Scholar] [CrossRef]
- Song, P.; Luo, M.; Liu, X.; Xing, W.; Xu, W.; Jiang, Z.; Gu, L. Zn Single Atom Catalyst for Highly Efficient Oxygen Reduction Reaction. Adv. Funct. Mater. 2017, 27, 1700802. [Google Scholar] [CrossRef]
- Wu, Z.-Y.; Xu, X.-X.; Hu, B.-C.; Liang, H.-W.; Lin, Y.; Chen, L.-F.; Yu, S.-H. Iron Carbide Nanoparticles Encapsulated in Mesoporous Fe-N-Doped Carbon Nanofibers for Efficient Electrocatalysis. Angew. Chem. Int. Ed. 2015, 54, 8179–8183. [Google Scholar] [CrossRef]
- Bai, L.; Hsu, C.-S.; Alexander, D.T.L.; Chen, H.M.; Hu, X. Double-Atom Catalysts as a Molecular Platform for Heterogeneous Oxygen Evolution Electrocatalysis. Nat. Energy 2021, 6, 1054–1066. [Google Scholar] [CrossRef]
- Kong, F.; Si, R.; Chen, N.; Wang, Q.; Li, J.; Yin, G.; Gu, M.; Wang, J.; Liu, L.-M.; Sun, X. Origin of Hetero-Nuclear Au-Co Dual Atoms for Efficient Acidic Oxygen Reduction. Appl. Catal. B Environ. 2022, 301, 120782. [Google Scholar] [CrossRef]
- Wei, B.; Fu, Z.; Legut, D.; Germann, T.C.; Du, S.; Zhang, H.; Francisco, J.S.; Zhang, R. Rational Design of Highly Stable and Active MXene-Based Bifunctional ORR/OER Double-Atom Catalysts. Adv. Mater. 2021, 33, 2102595. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, Q.; Wang, J.; Wang, J.; Zhang, J.; Zhao, Y. Supported Dual-Atom Catalysts: Preparation, Characterization, and Potential Applications. Chin. J. Catal. 2020, 41, 783–798. [Google Scholar] [CrossRef]
- Zhang, L.; Si, R.; Liu, H.; Chen, N.; Wang, Q.; Adair, K.; Wang, Z.; Chen, J.; Song, Z.; Li, J.; et al. Atomic Layer Deposited Pt-Ru Dual-Metal Dimers and Identifying Their Active Sites for Hydrogen Evolution Reaction. Nat. Commun. 2019, 10, 4936. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, Y.; Guo, X.; Chen, C.; Dong, C.-L.; Liu, R.-S.; Han, C.-P.; Li, Y.; Gogotsi, Y.; Wang, G. Single Platinum Atoms Immobilized on an MXene as an Efficient Catalyst for the Hydrogen Evolution Reaction. Nat. Catal. 2018, 1, 985–992. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and Simple Analytic Representation of the Electron-Gas Correlation Energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Moellmann, J.; Grimme, S. DFT-D3 Study of Some Molecular Crystals. J. Phys. Chem. C 2014, 118, 7615–7621. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A User-Friendly Interface Facilitating High-Throughput Computing and Analysis Using VASP Code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Wang, E.; Zhang, B.; Zhou, J.; Sun, Z. High Catalytic Activity of MBenes-Supported Single Atom Catalysts for Oxygen Reduction and Oxygen Evolution Reaction. Appl. Surf. Sci. 2022, 604, 154522. [Google Scholar] [CrossRef]
- Jing, T.; Liang, D.; Hao, J.; Deng, M.; Cai, S. Single Pt Atoms Stabilized on Mo2TiC2O2 for Hydrogen Evolution: A First-Principles Investigation. J. Chem. Phys. 2019, 151, 024702. [Google Scholar] [CrossRef]
- Kan, D.; Lian, R.; Wang, D.; Zhang, X.; Xu, J.; Gao, X.; Yu, Y.; Chen, G.; Wei, Y. Screening Effective Single-Atom ORR and OER Electrocatalysts from Pt Decorated MXenes by First-Principles Calculations. J. Mater. Chem. A 2020, 8, 17065–17077. [Google Scholar] [CrossRef]
- Huang, H.; Jia, H.; Liu, Z.; Gao, P.; Zhao, J.; Luo, Z.; Yang, J.; Zeng, J. Understanding of Strain Effects in the Electrochemical Reduction of CO2: Using Pd Nanostructures as an Ideal Platform. Angew. Chem. Int. Ed. 2017, 56, 3594–3598. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, E.; Guo, M.; Zhou, J.; Sun, Z. Reasonable Design of MXene-Supported Dual-Atom Catalysts with High Catalytic Activity for Hydrogen Evolution and Oxygen Evolution Reaction: A First-Principles Investigation. Materials 2023, 16, 1457. https://doi.org/10.3390/ma16041457
Wang E, Guo M, Zhou J, Sun Z. Reasonable Design of MXene-Supported Dual-Atom Catalysts with High Catalytic Activity for Hydrogen Evolution and Oxygen Evolution Reaction: A First-Principles Investigation. Materials. 2023; 16(4):1457. https://doi.org/10.3390/ma16041457
Chicago/Turabian StyleWang, Erpeng, Miaoqi Guo, Jian Zhou, and Zhimei Sun. 2023. "Reasonable Design of MXene-Supported Dual-Atom Catalysts with High Catalytic Activity for Hydrogen Evolution and Oxygen Evolution Reaction: A First-Principles Investigation" Materials 16, no. 4: 1457. https://doi.org/10.3390/ma16041457