

Eutectic In Situ Modification of Polyamide 12 Processed through Laser-Based Powder Bed Fusion

Abstract
:
1. Introduction
2. State of the Art
2.1. Thermal Material Properties in Quasi-Isothermal LPBF of Polymers
2.2. Properties of Eutectic Polymer-Based Compounds
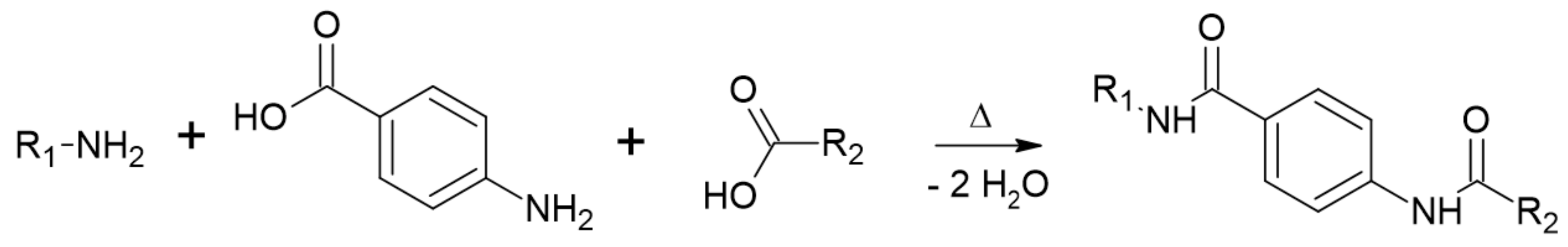
2.3. Synthesis Routes of Semi-Aromatic Polymers
2.4. Thermal Properties of Semi-Aromatic Polyamides
3. Materials and Methods
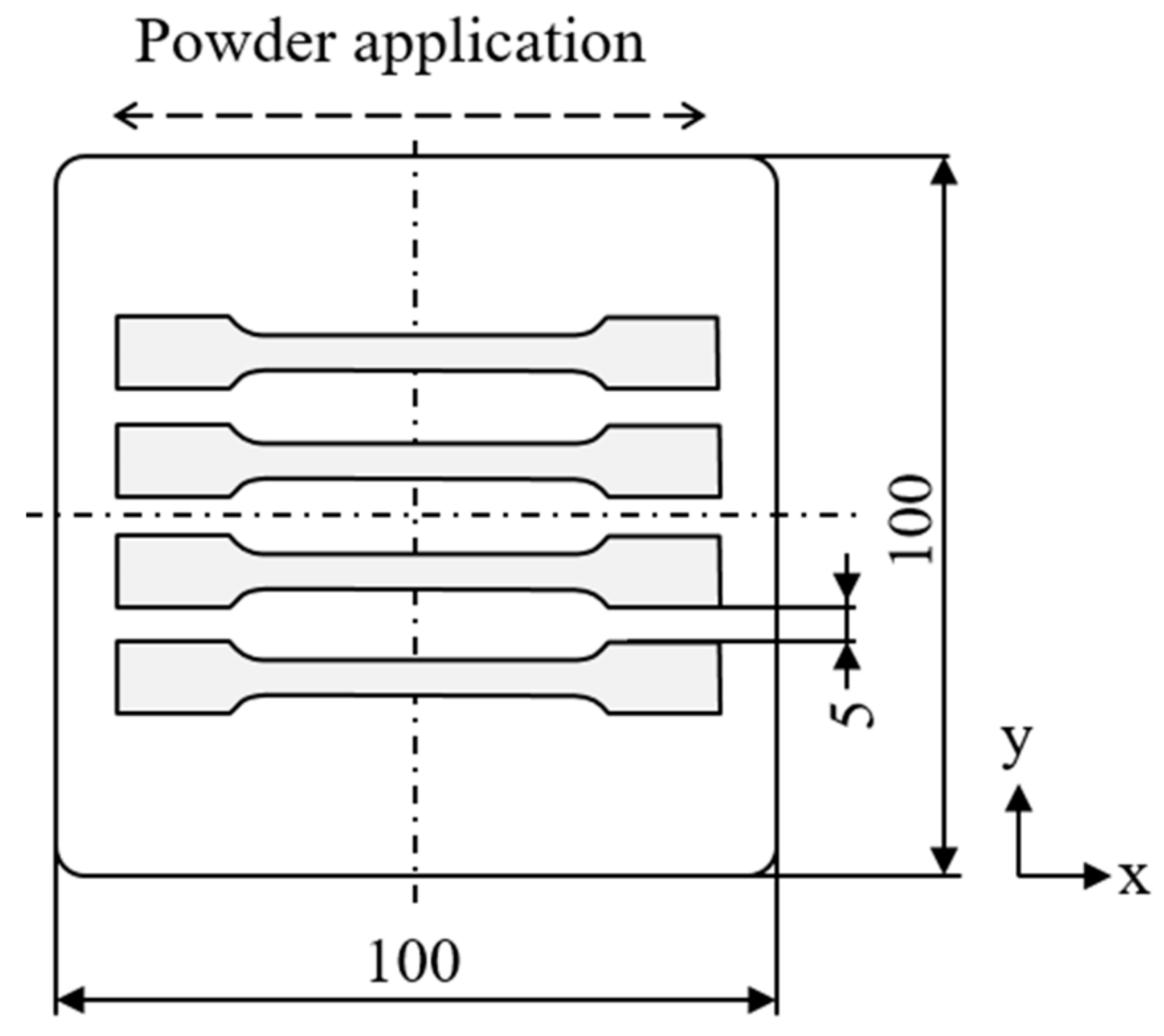
3.1. Material Preparation and Processing
3.2. Powder Blend Characterization
3.3. Characterization of Manufactured Components
4. Results and Discussion
4.1. Powder Blend Characteristics
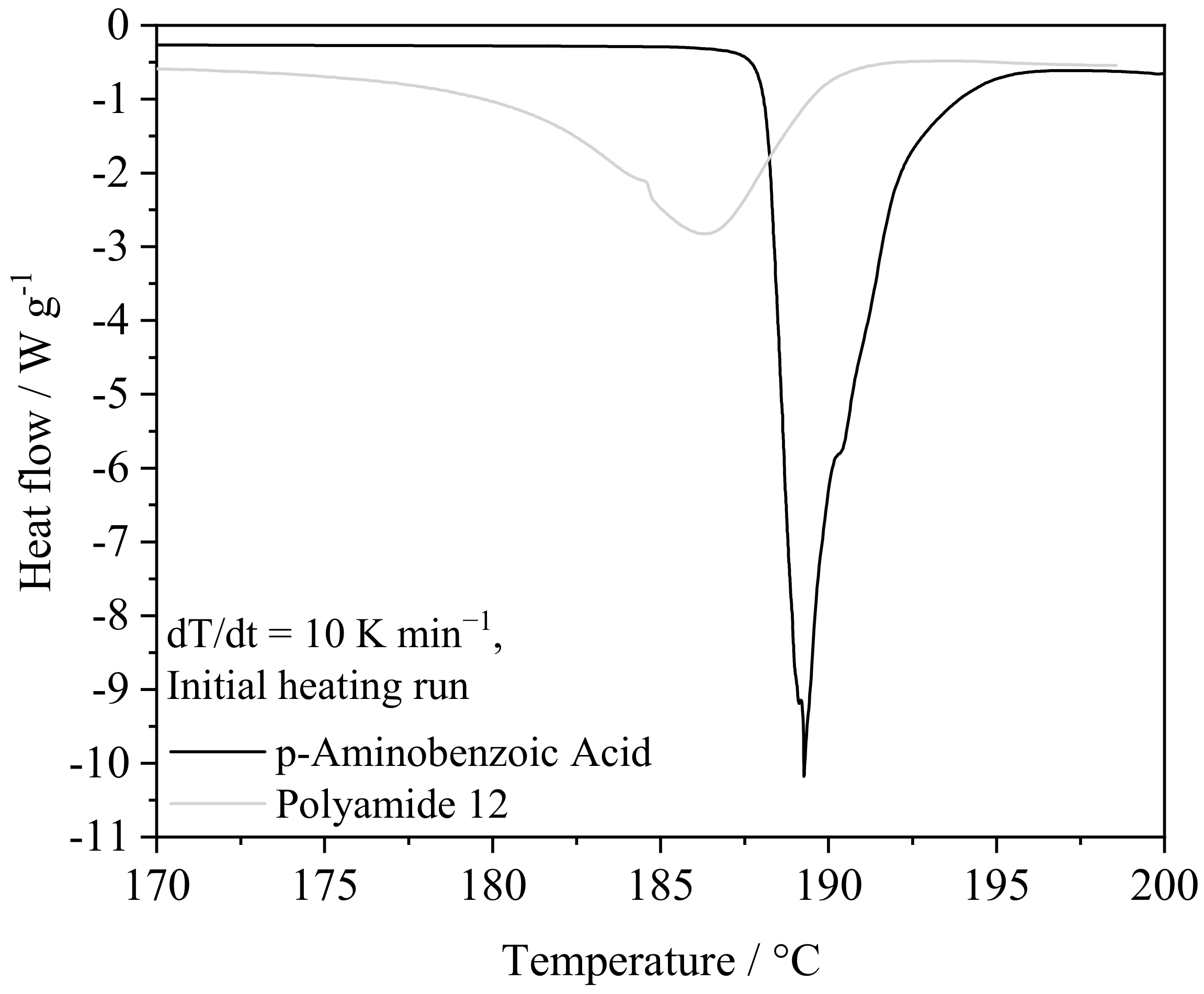
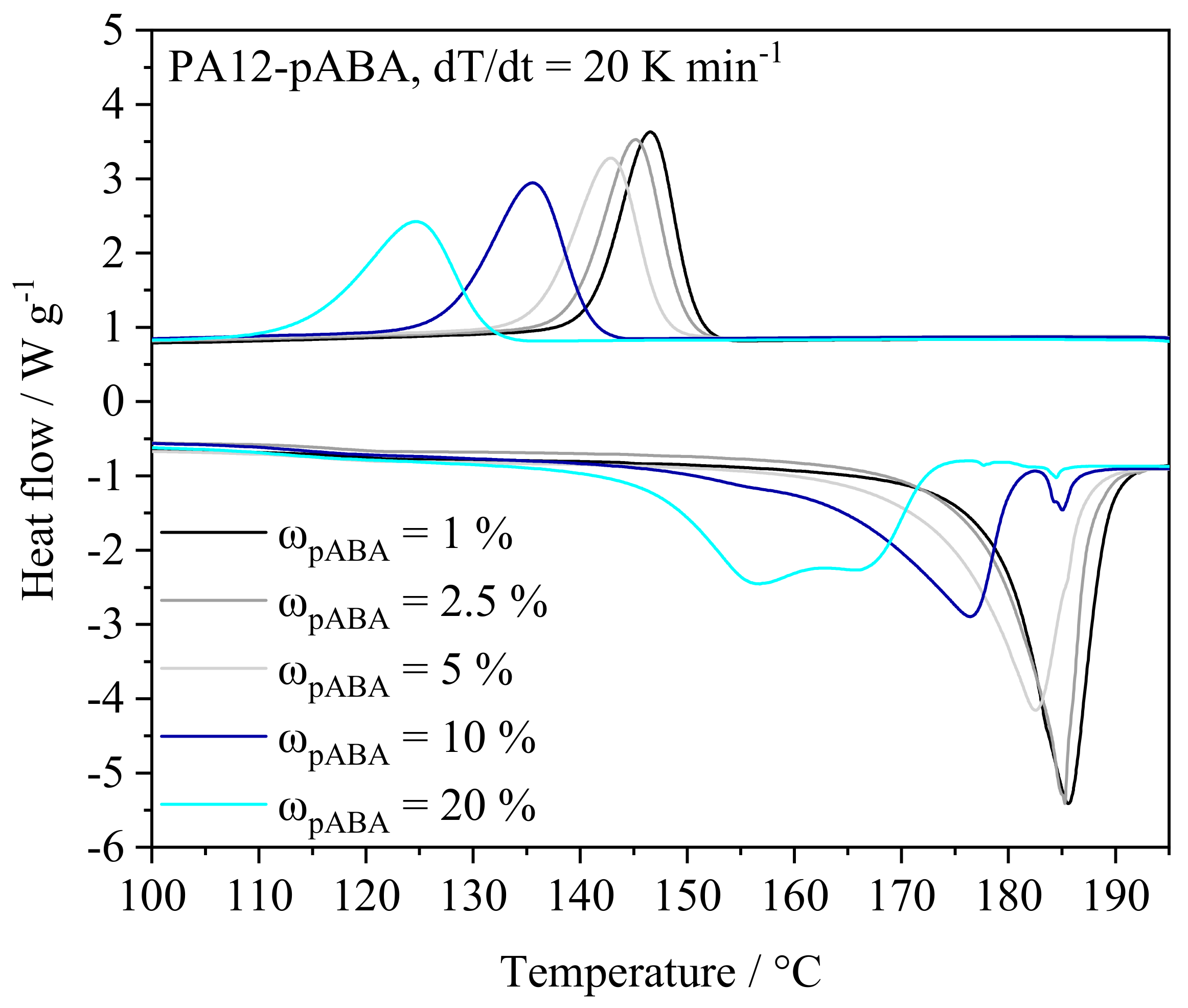
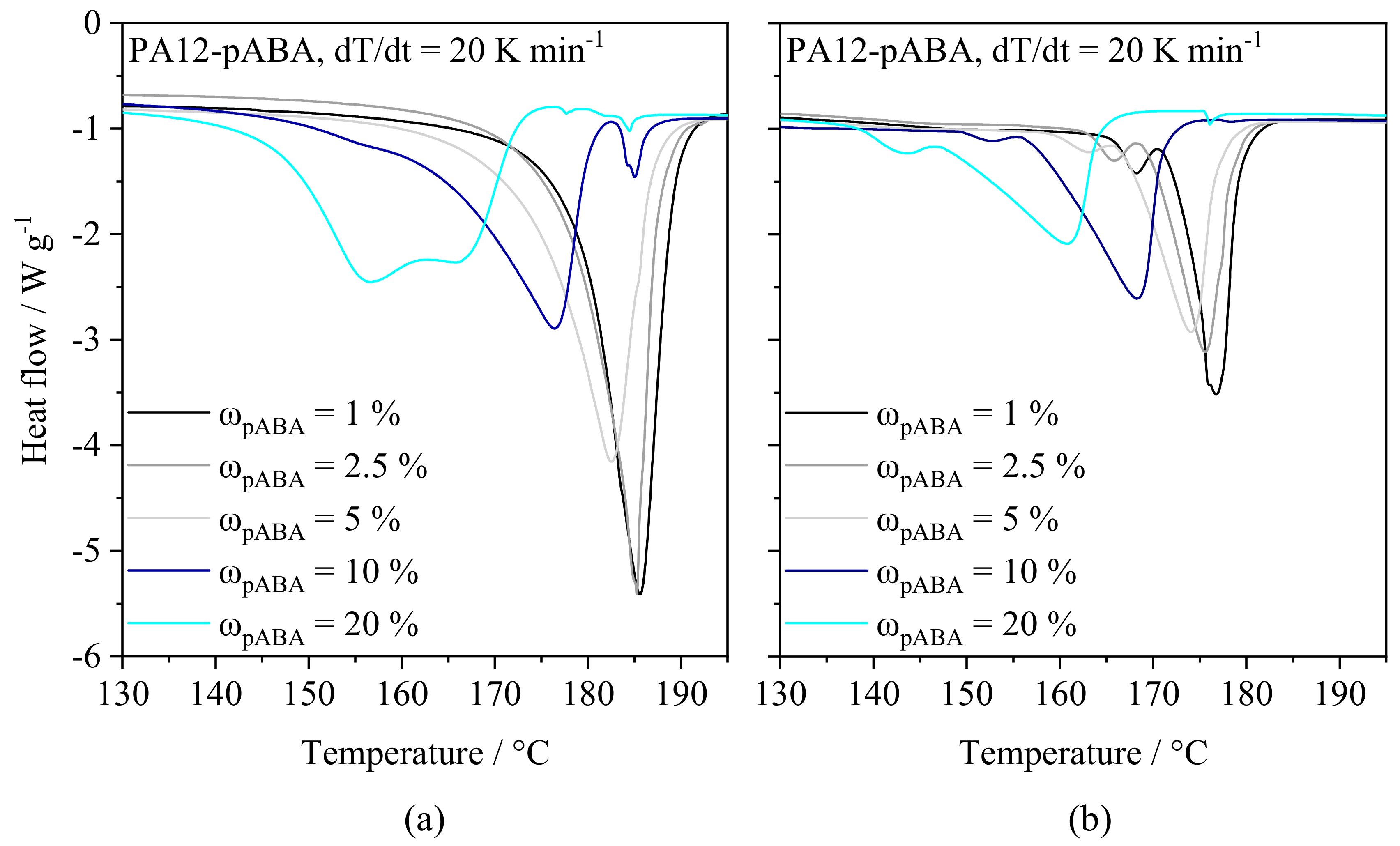
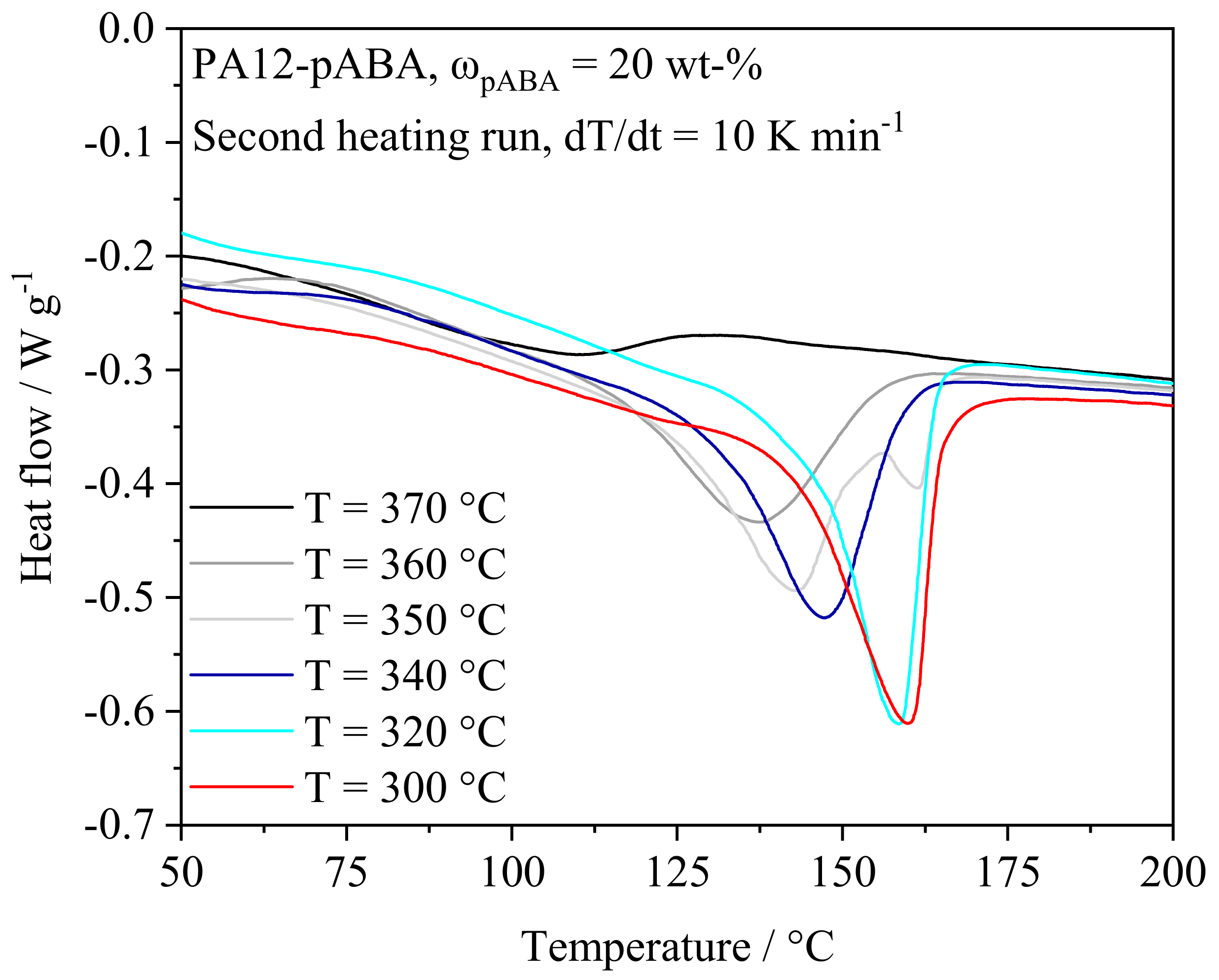
4.1.1. Thermal Powder Blend Properties
4.1.2. Rheological Powder Blend Properties
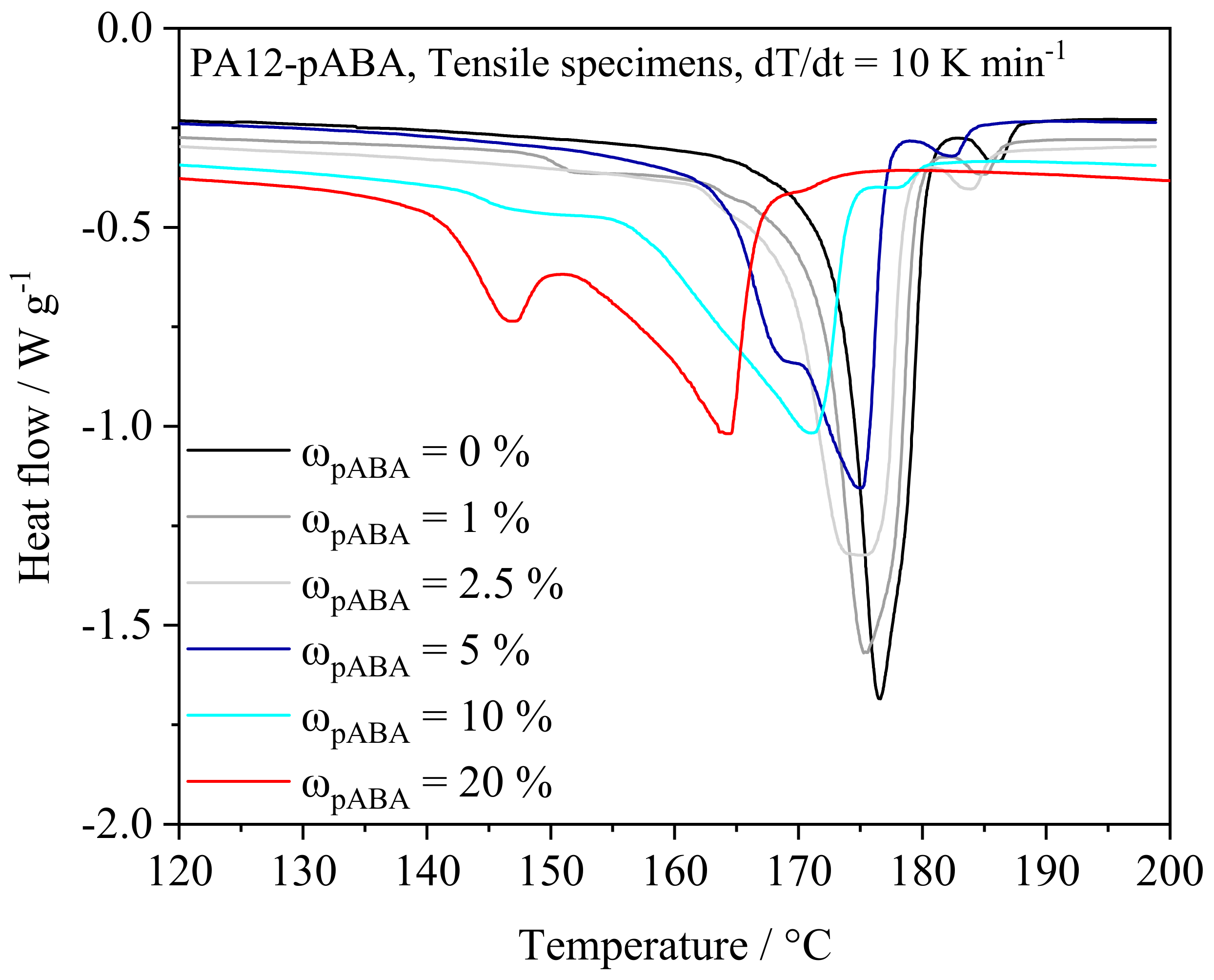
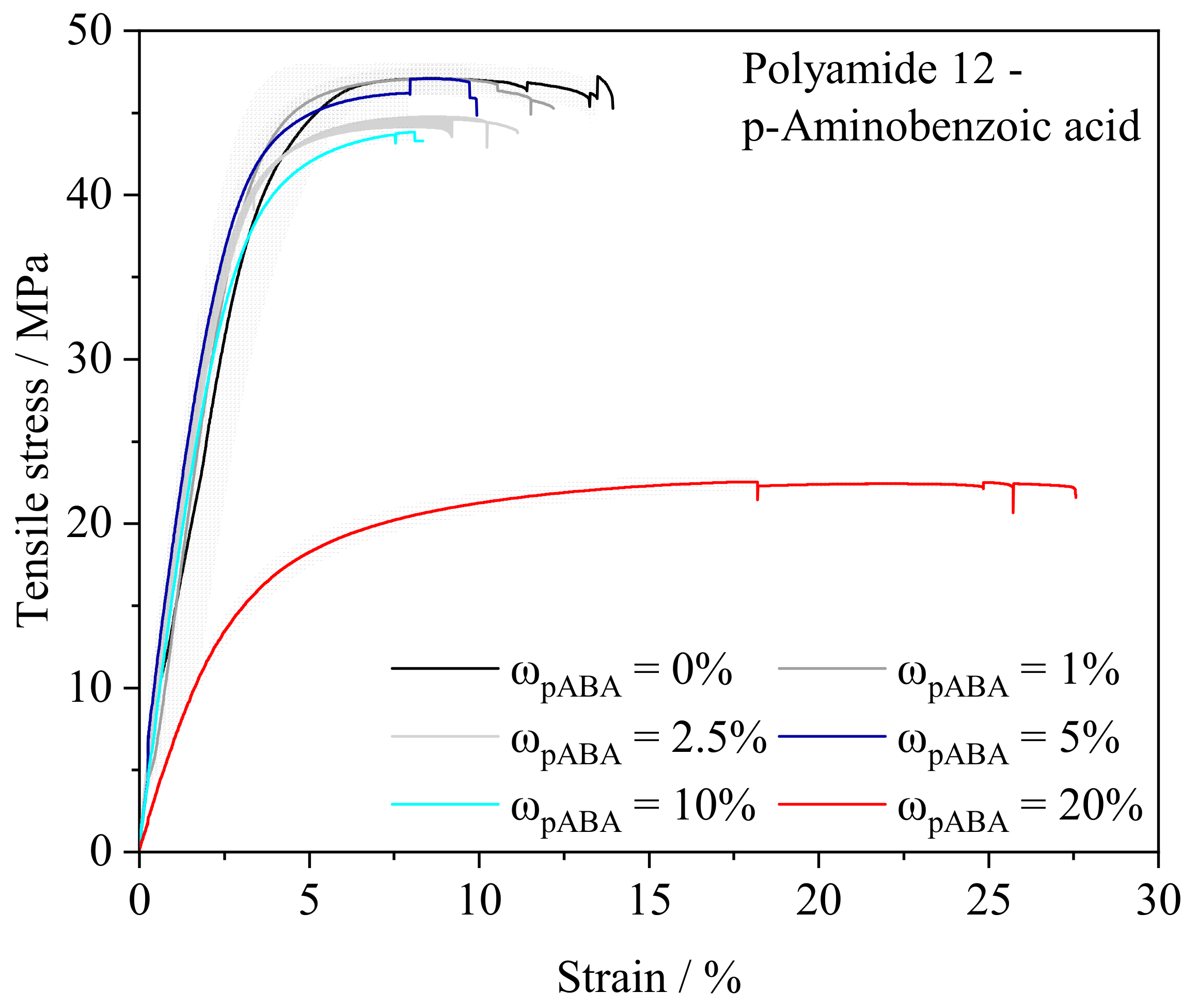
4.2. Mechanical and Thermal Part Properties
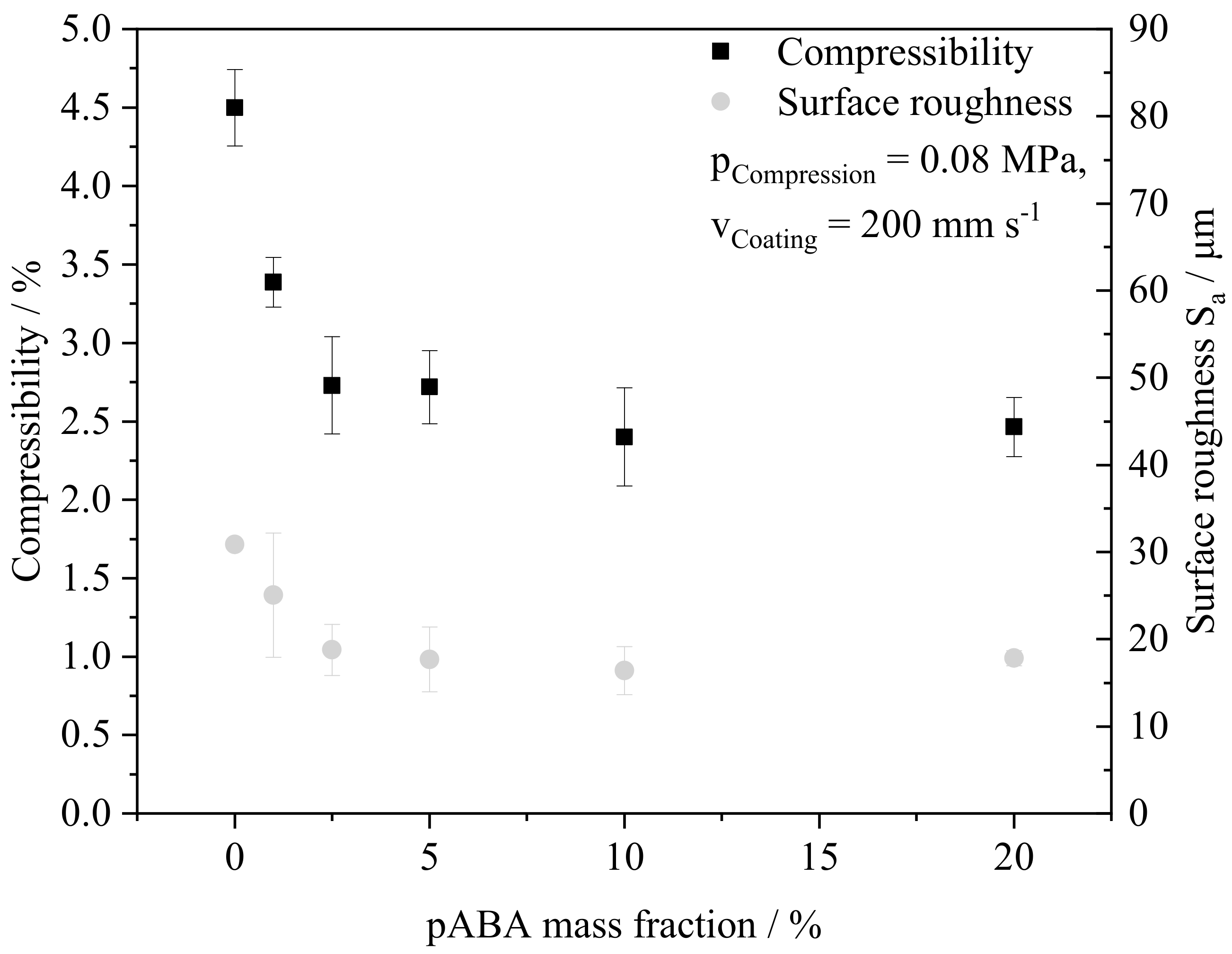
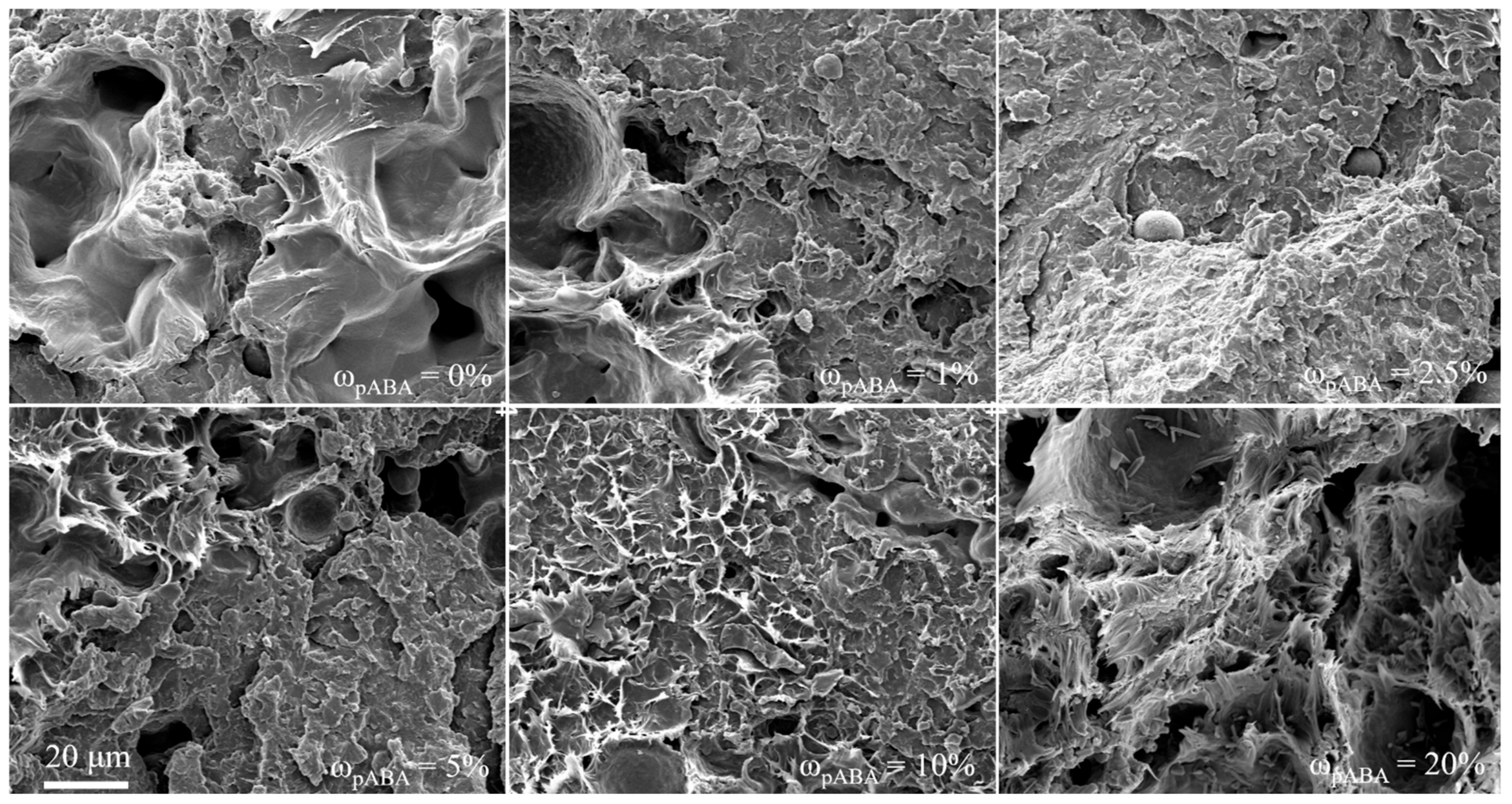
4.3. Part Morphology
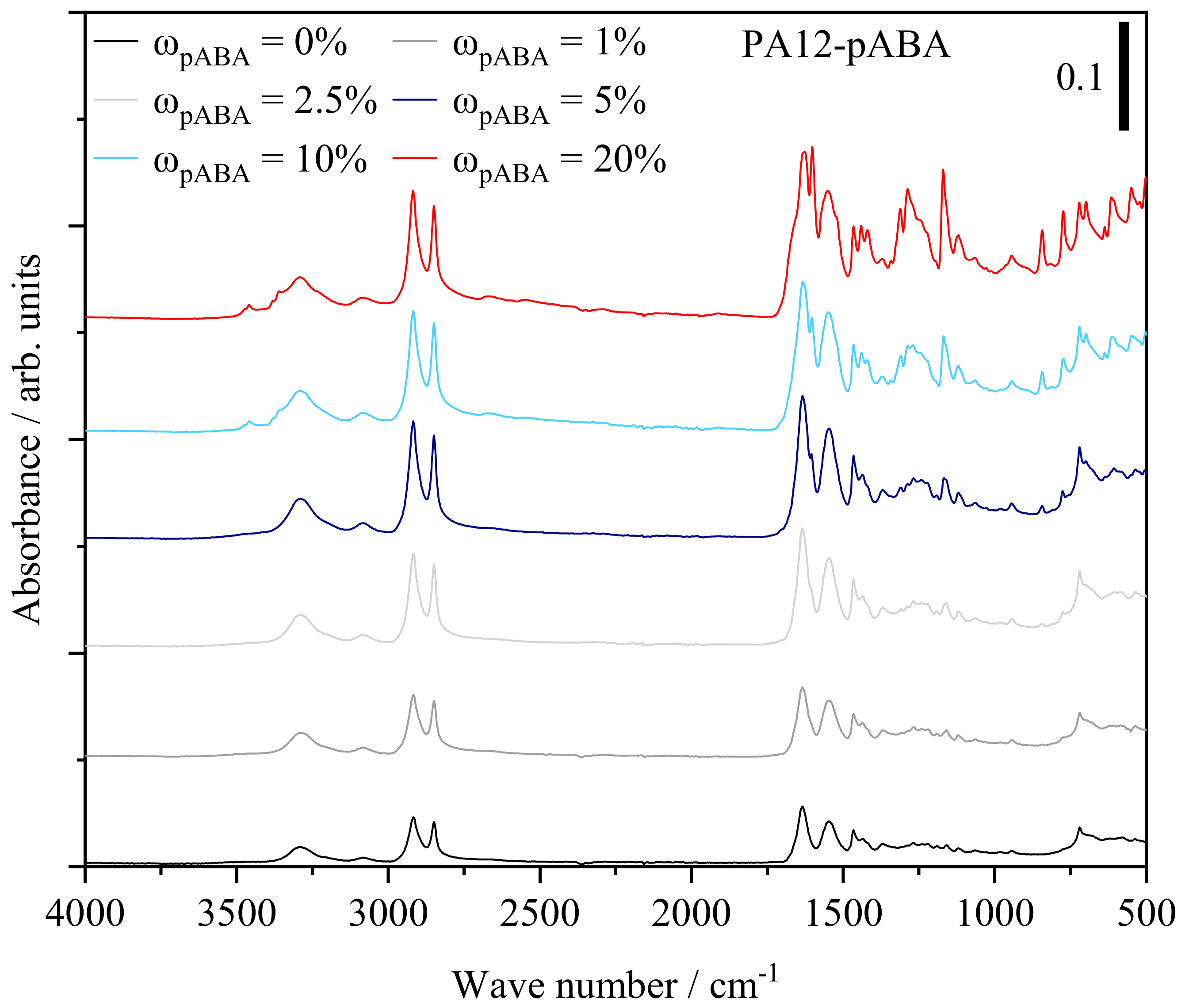
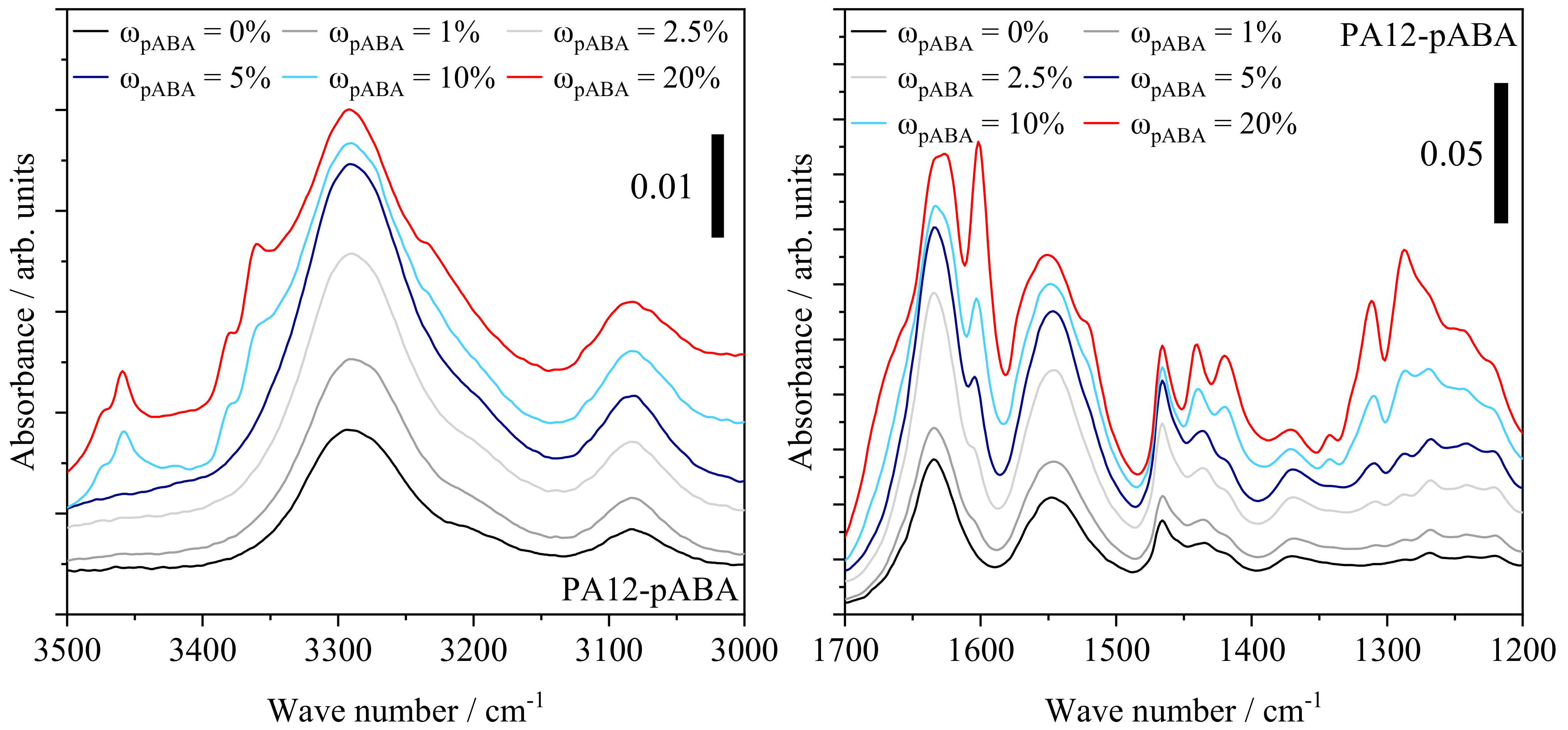
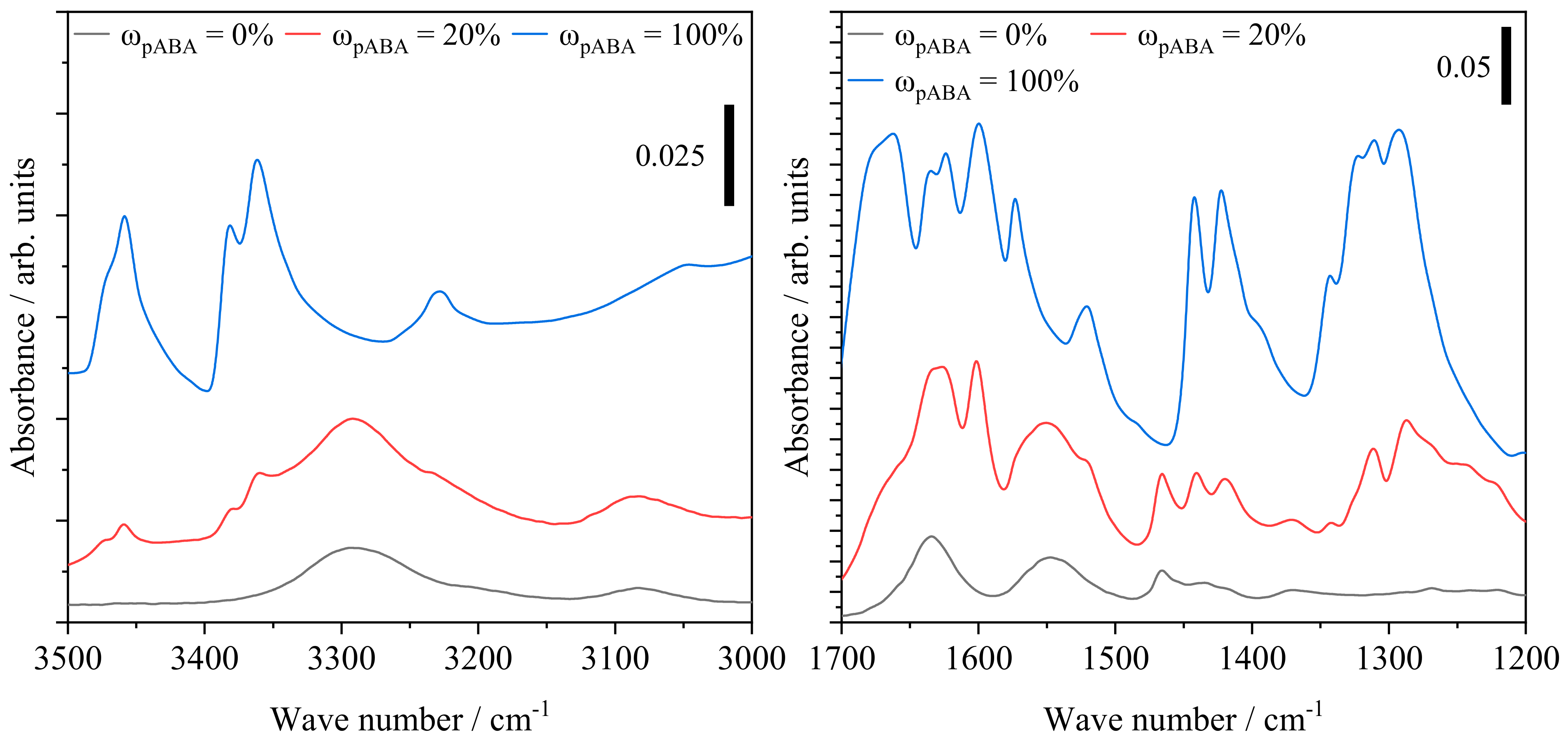
4.4. Qualitative Characterization of Covalent and Intermolecular Bonds
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rwei, S.-P.; Ranganathan, P.; Chiang, W.-Y.; Lee, Y.-H. Synthesis of Low Melting Temperature Aliphatic-Aromatic Copolyamides Derived from Novel Bio-Based Semi Aromatic Monomer. Polymers 2018, 10, 793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolffs, M.; Cotton, L.; Kolkman, A.J.; Rulkens, R. New sustainable alternating semi-aromatic polyamides prepared in bulk by direct solid-state polymerization. Polym. Int. 2021, 70, 546–554. [Google Scholar] [CrossRef]
- Peng, S.; Peng, L.; Yi, C.; Zhang, W.; Wang, X. A novel synthetic strategy for preparing semi-aromatic components modified polyamide 6 polymer. J. Polym. Sci. Part A Polym. Chem. 2018, 56, 959–967. [Google Scholar] [CrossRef]
- Higashihara, T.; Zhang, C.; Tsukuda, A.; Ochi, T.; Ueda, M. Direct synthesis and melt-drawing property of aramids by bulk polycondensation of isophthalic acid with m-phenylenediamine and 3,4′-oxydianiline. J. Appl. Polym. Sci. 2012, 124, 4398–4402. [Google Scholar] [CrossRef]
- Yamazaki, N.; Matsumoto, M.; Higashi, F. Studies on reactions of the N-phosphonium salts of pyridines. XIV. Wholly aromatic polyamides by the direct polycondensation reaction by using phosphites in the presence of metal salts. J. Polym. Sci. Polym. Chem. Ed. 1975, 13, 1373–1380. [Google Scholar] [CrossRef]
- Shibasaki, Y.; Abe, Y.; Sato, N.; Fujimori, A.; Oishi, Y. Direct condensation polymerization of N-alkylated p-aminobenzoic acid and packing of rigid-rod main chains with flexible side chains. Polym. J. 2010, 42, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Greiner, S.; Jaksch, A.; Cholewa, S.; Drummer, D. Development of material-adapted processing strategies for laser sintering of polyamide 12. Adv. Ind. Eng. Polym. Res. 2021, 4, 251–263. [Google Scholar] [CrossRef]
- Drummer, D.; Greiner, S.; Zhao, M.; Wudy, K. A novel approach for understanding laser sintering of polymers. Addit. Manuf. 2019, 27, 379–388. [Google Scholar] [CrossRef]
- Soldner, D.; Greiner, S.; Burkhardt, C.; Drummer, D.; Steinmann, P.; Mergheim, J. Numerical and experimental investigation of the isothermal assumption in selective laser sintering of PA12. Addit. Manuf. 2020, 37, 101676. [Google Scholar] [CrossRef]
- Soldner, D.; Steinmann, P.; Mergheim, J. Modeling crystallization kinetics for selective laser sintering of polyamide 12. GAMM-Mitteilungen 2021, 44, e202100011. [Google Scholar] [CrossRef]
- Wudy, K.; Drummer, D. Aging effects of polyamide 12 in selective laser sintering: Molecular weight distribution and thermal properties. Addit. Manuf. 2019, 25, 1–9. [Google Scholar] [CrossRef]
- El-Mazry, C.; Ben Hassine, M.; Correc, O.; Colin, X. Thermal oxidation kinetics of additive free polyamide 6-6. Polym. Degrad. Stab. 2013, 98, 22–36. [Google Scholar] [CrossRef] [Green Version]
- Pliquet, M.; Rapeaux, M.; Delange, F.; Bussiere, P.O.; Therias, S.; Gardette, J.L. Multiscale analysis of the thermal degradation of polyamide 6,6: Correlating chemical structure to mechanical properties. Polym. Degrad. Stab. 2021, 185, 109496. [Google Scholar] [CrossRef]
- Lanzl, L.; Wudy, K.; Greiner, S.; Drummer, D. Selective laser sintering of copper filled polyamide 12: Characterization of powder properties and process behavior. Polym. Compos. 2019, 40, 1801–1809. [Google Scholar] [CrossRef]
- Lanzl, L.; Wudy, K.; Drummer, D. The effect of short glass fibers on the process behavior of polyamide 12 during selective laser beam melting. Polym. Test. 2020, 83, 106313. [Google Scholar] [CrossRef]
- Roda, A.; Matias, A.A.; Paiva, A.; Duarte, A.R.C. Polymer Science and Engineering Using Deep Eutectic Solvents. Polymers 2019, 11, 912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janicka, P.; Kaykhaii, M.; Płotka-Wasylka, J.; Gębicki, J. Supramolecular deep eutectic solvents and their applications. Green Chem. 2022, 24, 5035–5045. [Google Scholar] [CrossRef]
- Hansen, B.B.; Spittle, S.; Chen, B.; Poe, D.; Zhang, Y.; Klein, J.M.; Horton, A.; Adhikari, L.; Zelovich, T.; Doherty, B.W.; et al. Deep Eutectic Solvents: A Review of Fundamentals and Applications. Chem. Rev. 2021, 121, 1232–1285. [Google Scholar] [CrossRef]
- Stefanovic, R.; Ludwig, M.; Webber, G.B.; Atkin, R.; Page, A.J. Nanostructure, hydrogen bonding and rheology in choline chloride deep eutectic solvents as a function of the hydrogen bond donor. Phys. Chem. Chem. Phys. 2017, 19, 3297–3306. [Google Scholar] [CrossRef]
- Ibrahim, R.K.; Hayyan, M.; AlSaadi, M.A.; Ibrahim, S.; Hayyan, A.; Hashim, M.A. Physical properties of ethylene glycol-based deep eutectic solvents. J. Mol. Liq. 2019, 276, 794–800. [Google Scholar] [CrossRef]
- Abbott, A.P.; Capper, G.; Davies, D.L.; Rasheed, R.K.; Tambyrajah, V. Novel solvent properties of choline chloride/urea mixtures. Chem. Commun. 2003, 70–71. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Cai, C.; Li, F.; Tan, Z.; Dong, S. Deep Eutectic Supramolecular Polymers: Bulk Supramolecular Materials. Angew. Chem. Int. Ed. 2020, 59, 11871–11875. [Google Scholar] [CrossRef] [PubMed]
- El Achkar, T.; Moufawad, T.; Ruellan, S.; Landy, D.; Greige-Gerges, H.; Fourmentin, S. Cyclodextrins: From solute to solvent. Chem. Commun. 2020, 56, 3385–3388. [Google Scholar] [CrossRef] [PubMed]
- De Lacalle, J.L.; Gallastegui, A.; Olmedo-Martínez, J.L.; Moya, M.; Lopez-Larrea, N.; Picchio, M.L.; Mecerreyes, D. Multifunctional Ionic Polymers from Deep Eutectic Monomers Based on Polyphenols. ACS Macro Lett. 2023, 12, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, J.C.; Manley, R.S.J. Polymer–monomer binary mixtures. I. Eutectic and epitaxial crystallization in poly (ε-caprolactone)–trioxane mixtures. J. Polym. Sci. Polym. Phys. Ed. 1977, 15, 1089–1100. [Google Scholar] [CrossRef]
- Papaspyrides, C.D.; Porfyris, A.D.; Vouyiouka, S.; Rulkens, R.; Grolman, E.; Poel, G.V. Solid state polymerization in a micro-reactor: The case of poly (tetramethylene terephthalamide). J. Appl. Polym. Sci. 2016, 133, 14. [Google Scholar] [CrossRef]
- Zhang, C.; Shoji, Y.; Higashihara, T.; Tsukuda, A.; Ochi, T.; Ueda, M. Synthesis of poly (m-phenyleneisophthalamide) by solid-state polycondensation of isophthalic acid with m-phenylenediamine. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 4725–4728. [Google Scholar] [CrossRef]
- Jeyakumar, A.; Goossens, H.; Noordover, B.; Prusty, M.; Scheibitz, M.; Koning, C. Polyamide-6,6-based blocky copolyamides obtained by solid-state modification. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 5118–5129. [Google Scholar] [CrossRef]
- Papaspyrides, C.D.; Vouyiouka, S.N.; Bletsos, I.V. New aspects on the mechanism of the solid state polyamidation of PA 6,6 salt. Polymer (Guildf). 2006, 47, 1020–1027. [Google Scholar] [CrossRef]
- Papaspyrides, C.D.; Porfyris, A.D.; Rulkens, R.; Grolman, E.; Kolkman, A.J. The effect of diamine length on the direct solid state polycondensation of semi-aromatic nylon salts. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 2493–2506. [Google Scholar] [CrossRef]
- Porfyris, A.; Vouyiouka, S.; Papaspyrides, C.; Rulkens, R.; Grolman, E.; Vanden Poel, G. Investigating alternative routes for semi-aromatic polyamide salt preparation: The case of tetramethylenediammonium terephthalate (4T salt). J. Appl. Polym. Sci. 2016, 133, 13. [Google Scholar] [CrossRef]
- Zhang, C. Progress in semicrystalline heat-resistant polyamides. e-Polymers 2018, 18, 373–408. [Google Scholar] [CrossRef]
- Edgar, O.B.; Hill, R. The p-phenylene linkage in linear high polymers: Some structure–property relationships. J. Polym. Sci. 1952, 8, 1–22. [Google Scholar] [CrossRef]
- Endo, T.; Higashihara, T. Direct Synthesis of Thermally Stable Semiaromatic Polyamides by Bulk Polymerization Using Aromatic Diamines and Aliphatic Dicarboxylic Acids. ACS Omega 2022, 7, 8753–8758. [Google Scholar] [CrossRef]
- Hesse, N.; Winzer, B.; Peukert, W.; Schmidt, J. Towards a generally applicable methodology for the characterization of particle properties relevant to processing in powder bed fusion of polymers–From single particle to bulk solid behavior. Addit. Manuf. 2021, 41, 101957. [Google Scholar] [CrossRef]
- Sillani, F.; Schiegg, R.; Schmid, M.; MacDonald, E.; Wegener, K. Powder Surface Roughness as Proxy for Bed Density in Powder Bed Fusion of Polymers. Polymers 2022, 14, 81. [Google Scholar] [CrossRef]
- Gueche, Y.A.; Sanchez-Ballester, N.M.; Bataille, B.; Aubert, A.; Leclercq, L.; Rossi, J.-C.; Soulairol, I. Selective Laser Sintering of Solid Oral Dosage Forms with Copovidone and Paracetamol Using a CO (2) Laser. Pharmaceutics 2021, 13, 160. [Google Scholar] [CrossRef]
- Beitz, S.; Uerlich, R.; Bokelmann, T.; Diener, A.; Vietor, T.; Kwade, A. Influence of Powder Deposition on Powder Bed and Specimen Properties. Materials 2019, 12, 297. [Google Scholar] [CrossRef] [Green Version]
- Benz, J.; Bonten, C. Temperature induced ageing of PA12 powder during selective laser sintering process. AIP Conf. Proc. 2019, 2055, 140001. [Google Scholar] [CrossRef]
- Drummer, D.; Wudy, K.; Drexler, M. Modelling of the aging behavior of polyamide 12 powder during laser melting process. AIP Conf. Proc. 2015, 1664, 160007. [Google Scholar] [CrossRef]
- Jeyakumar, A. Solid-State Modification of Polyamide-6,6. Ph.D. Thesis, Technische Universiteit Eindhoven, Eindhoven, The Netherlands, 2012. [Google Scholar]
- Guo, L.; Sato, H.; Hashimoto, T.; Ozaki, Y. FTIR Study on Hydrogen-Bonding Interactions in Biodegradable Polymer Blends of Poly (3-hydroxybutyrate) and Poly (4-vinylphenol). Macromolecules 2010, 43, 3897–3902. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pABA Mass Fraction/% | pABA Molar Fraction/mol.-% | Isothermal Build Chamber Temperature/°C |
|---|---|---|
| 0.0 | 0.00 | 172.0 |
| 1.0 | 1.56 | 170.0 |
| 2.5 | 3.87 | 167.5 |
| 5.0 | 7.63 | 155.0 |
| 10 | 14.86 | 144.5 |
| 20 | 28.19 | 141.5 |
| Isothermal Temperature/°C | Isothermal Reaction Time/s |
|---|---|
| 300 | 600 |
| 320 | 600 |
| 340 | 600 |
| 350 | 600 |
| 360 | 600 |
| 370 | 600 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schlicht, S.; Drummer, D. Eutectic In Situ Modification of Polyamide 12 Processed through Laser-Based Powder Bed Fusion. Materials 2023, 16, 2050. https://doi.org/10.3390/ma16052050
Schlicht S, Drummer D. Eutectic In Situ Modification of Polyamide 12 Processed through Laser-Based Powder Bed Fusion. Materials. 2023; 16(5):2050. https://doi.org/10.3390/ma16052050
Chicago/Turabian StyleSchlicht, Samuel, and Dietmar Drummer. 2023. "Eutectic In Situ Modification of Polyamide 12 Processed through Laser-Based Powder Bed Fusion" Materials 16, no. 5: 2050. https://doi.org/10.3390/ma16052050