The production of 1,2-PG has been extensively studied in the literature. Bulk and supported catalysts based on different metal phases have been used, with the particle size and the surface acidity being the relevant catalytic properties in the production of this compound.
Regarding the bulk catalysts, Cu, Ni, and Co have been used due to their low cost compared to noble metals. Of all of them, Cu-based bulk catalysts were the first to be studied due to the ability of Cu to cleave C-O bonds. Within the set of supported catalysts, Ru, Pt, and Pd metal phases have been used, in addition to the previously mentioned non-noble metals.
Traditional supports based on metallic and non-metallic oxides as well as carbonaceous supports have been studied under the reaction conditions. Regardless of the metal phase, the acid-base properties of the supports are key to obtain high selectivity values at 1,2-PG.
The most relevant aspects in the study of catalysts used in the hydrogenolysis reaction of glycerol to 1,2-PG are described below, differentiating according to the metal phase and the type of reactor used, both in liquid and vapor phase.
3.1. Ru Catalysts
Of the set of supported metal catalysts for the production of 1,2-PG, those based on Ru as the active phase have been the most studied ones due to the high intrinsic activity of the metal.
Table 1 summarizes the operating conditions and best activity results of supported Ru catalysts in batch reactors. In all cases, the maximum performance achieved at 1,2-PG is reported.
Dasari et al. were the first authors to evaluate commercial Ru/C and Ru/Al
2O
3 catalysts using 80% aqueous glycerol solutions. The results showed that both catalysts were active, but not as selective to 1,2-PG as the Cu catalysts (~40% for Ru/C and ~60% for Ru/Al
2O
3). The authors assigned these results to the lower ability of Ru to hydrogenate C-O bonds compared to the Cu metal phase [
45]. Following these results, Maris et al. studied commercial Ru/C catalysts and concluded that Ru promotes C-C bond cleavage reactions, favoring the formation of side products such as EG and EtOH [
43].
With respect to other noble metals (Pt, Pd, Rh), Ru has been shown to have a higher intrinsic activity in the hydrogenolysis of glycerol. Maris et al. compared the activity of Ru/C with the activity of Pt/C and concluded that the activity of the former is higher than that of the latter under the same operating conditions [
43]. Alhanash et al. evaluated Ru and Rh catalysts supported on a heteropolyacid salt (CsPW). Ru/CsPW proved to be more active than Rh/CsPW, allowing to obtain high selectivity to 1,2-PG (96%) with glycerol conversions of 20%, employing low hydrogen pressure (0.5 MPa) [
79]. Wang et al. demonstrated that the Ru/ZrO
2 catalyst was the most active one compared to Rh, Pt, and Pd catalysts. The order of activity, for particle sizes of the order of 2 nm, was as follows: Ru > Rh > Pt > Pd [
87].
Some articles reported that the activity of Ru-supported catalysts is affected by the metal particle size and the acidity of the catalyst. With respect to particle size, it has been reported that small particle sizes result in high dispersions that increase conversion levels. In addition, it has been reported that the particle size depends on the type of the Ru precursor, the preparation method, and the support. On the other hand, the surface properties of the support have the greatest impact on the acidity of the catalyst.
The most commonly employed Ru precursors have been RuCl
3 [
48,
53,
72,
73,
74,
75,
76,
77,
79,
80,
81,
85,
86,
87,
88,
89,
90,
91,
92], Ru(NO)(NO
3)
3 [
48,
77,
84,
86,
91], Ru (AcAc)
3 [
48,
82], and Ru
3(CO)
12 [
82].
Several studies have reported that of all the precursors, Ru(NO)(NO
3)
3 allows for obtaining the highest glycerol conversion and selectivity to 1,2-PG [
48,
77,
86,
91,
92,
93] because its decomposition generates small Ru particles with sizes smaller than 2 nm [
48]. A similar effect was reported employing RuCl
3, which has been one of the most widely used precursors [
93]. However, some articles have highlighted that the presence of Cl
− ions generate acidic Bronsted sites on the surface of the supports [
72] that would be responsible for promoting the formation of 1-POH, 2-POH [
77], EG, and MeOH [
86]. Other precursors such as Ru(AcAc)
3 and Ru
3(CO)
12 have been less employed and in comparison to the other precursors have shown lower levels of activity [
48,
82]. Based on these results, the following order of activity per precursor type could be established:
With respect to the preparation methods, the most commonly employed have been the wetness incipient impregnation [
43,
48,
72,
73,
75,
77,
79,
80,
81,
82,
84,
85,
86,
87,
88,
90,
93] and deposition–precipitation [
76,
90]. Balaraju et al. prepared Ru/TiO
2 catalysts employing both methods. Their results showed that the deposition–precipitation method allowed higher metal dispersion (29–53%) to be achieved, due to the reduction in Ru(OH)
3 species generated during the catalyst preparation process, which led to obtaining small size metal particles (dva ~ 1–3 nm). With the use of the incipient moisture impregnation method, the formation of larger Ru particles (dva ~ 10 nm) led to lower activity levels [
90].
Catalyst activation has also been indicated as a variable to consider in metal particle size. For Ru/C [
48] and Ru/TiO
2 [
81] catalysts, it has been reported that high activation temperatures lead to the agglomeration of Ru particles, and this causes a drop in conversion levels. For Ru/AC catalysts, the reduction in the H
2 atmosphere was found to be less effective than the reduction in the presence of NaBH
4 in the liquid phase due to the larger Ru particle sizes obtained (3–5 nm) [
86].
The support also impacts on the activity levels, not only because of its effect on Ru particle size but also because of its acid-base properties. Reports have indicated the use of carbonaceous supports in the form of carbon graphite (C), activated carbons (AC), carbon nanotubes (CNT), and high specific surface-area graphite (HSAG), in addition to oxides such as Al
2O
3, SiO
2, ZrO
2, and TiO
2, and zeolites of the Hβ and HZSM-5 type. Within this set, carbonaceous supports have been the most studied ones, because they provide the necessary specific surface area for the formation of Ru particles with high dispersion [
72,
78,
92] and can be functionalized to increase their acidity [
91].
With respect to the effect of the particle size, activated carbons (AC) have proved to be better supports than graphitic carbons (C) with a different degree of crystallinity, because they led to a higher dispersion of Ru. Mane et al. indicated that Ru/AC was the most active and selective catalyst to 1,2-PG due to the formation of Ru particles with sizes between 1.1 and 1.7 nm. The use of Ru/C catalysts with Ru particles of larger size, between 4 and 5 nm, and with the presence of Ru oxides, presented lower levels of activity [
92]. On the other hand, Ru catalysts supported on CNT and HSAG proved to be more active than Ru/AC catalysts under the same reaction conditions, due to the formation of a higher amount of electron-rich Ru
δ− species that favored the cleavage of the terminal C-O bond of glycerol to form 1,2-PG [
72].
With respect to acidity, the carbonaceous supports would have the necessary acidic properties in the hydrogenolysis reaction. However, a specific type of acid site is required, and the number of sites must be appropriate to avoid losses in selectivity to 1,2-PG. Gallegos-Suarez et al. studied Ru/AC catalysts modified by functionalization with nitric acid. The results showed that -COOH groups, generated during carbon oxidation, favor the cleavage of C-O bonds. In addition, the study showed that, although the activity was promoted, the selectivity to 1,2-PG decreased compared to catalysts supported on unfunctionalized AC. The results were attributed to the high level of acidity provided by the functionalization process which resulted in the formation of side products such as 1-POH, 2-POH, MeOH and other gases such as methane, ethane, and propane [
91].
Some traditional oxides such as Al
2O
3, SiO
2, ZrO
2, TiO
2, and Hβ-type and HZSM-5 zeolites were also employed as supports, but the activity of Ru on these supports was lower than the results obtained employing carbonaceous ones. Ma et al. reported the use of Al
2O
3 and ZrO
2 as supports [
88], as well as SiO
2, TiO
2, and those of the Hβ and HZSM-5 type [
74]. The order of activity (based on the yield to 1,2-PG) was as follows:
The authors found that the Ru particle size is smaller for the Ru/C catalyst, followed by the Ru/ZrO
2 and Ru/SiO
2 catalysts, suggesting that the activity found is due to the higher dispersion obtained on the supports [
88]. However, the activity for the zeolite-based catalysts also showed good activity, suggesting that the role of acidity is not less important in such materials.
Other less-studied supports were hydrotalcites. Lee et al. prepared Ru catalysts supported on a CaMgZn-Al type hydrotalcite. The results showed high levels of conversion and selectivity to 1,2-PG due to the higher acidity of the hydrotalcite with respect to other supports, such as γ-Al
2O
3, and the smaller metal particle size (d
va ~ 13.7 nm) [
84].
With the aim of improving the surface textural and acidic properties, the modification of some of the traditional oxides used as supports has been studied.
For the ZrO
2 support, the addition of WO
3 led to a decrease in Ru/ZrO
2 catalyst activity, but an improvement in selectivity to 1,2-PG, due to the suppression of C-C bond cleavage reactions [
73]. The addition of La to the same support, on the other hand, caused a decrease in selectivity to 1,2-PG due to the promotion of C-C bond cleavage reactions, that increased the selectivity to EG [
76].
For the TiO
2 support, it has been demonstrated that it is possible to obtain good metallic dispersions by modification with clays. Hamzah et al. employed a TiO
2: bentonite support (with a 1:2 mass ratio) and obtained Ru particles of the order of 1.5 nm that allowed the achievement of a high glycerol conversion (~70%) and selectivity to 1,2-PG (~80%) [
85].
With respect to Ru/Al
2O
3 catalysts, the modification of the support with AlF
3 for contents between 17 and 58 wt.% allowed the maximum glycerol conversions to be reached, due to a higher dispersion of Ru particles; however, it also promoted the formation of EG and gases such as methane [
75], probably due to the surface acidity generated by the presence of the modifier.
Other more complex catalytic systems, such as Ru/LaCO
3OH, were found to be more active, selective, and stable than the Ru/SiO
2 and Ru/ZrO
2 catalysts. Ru/LaCO
3OH was prepared from the precursor RuCl
3, which reacts during impregnation with the support to form a complex of LaRu(CO
3)
2Cl
2 and LaOCl. During hydrothermal reduction, these intermediates hydrolyze to form LaCO
3OH and Ru(OH)
3, which subsequently transform into Ru0 nanoparticles. The improvement in activity levels is due to the presence of Ru0 encapsulated by a protective layer of LaCO
3OH [
80].
With the aim of increasing the glycerol conversion and improving the selectivity to 1,2-PG, bimetallic Ru catalysts have been studied employing several metals, such as Pt [
94,
95], Au [
94], Re [
74,
88,
96,
97,
98], Cu [
99,
100], Co [
100,
101,
102], Ni [
100] and Fe [
103].
Table 2 summarizes the operating conditions and activity results of Ru bimetallic catalysts in batch reactors.
Some articles have reported that Ru–Pt and Ru–Au combinations did not produce improvements in the activity of monometallic Ru catalysts. The Ru–Pt/C combination, for example, did not generate an increase in the levels of conversion and selectivity to 1,2-PG with respect to a Ru/C catalyst, while the Ru–Au combination produced a higher selectivity to EG and lactate, decreasing the selectivity values to 1,2-PG. Moreover, the incorporation of Au favored the deactivation of the catalyst due to the sintering of the Ru particles [
94].
On the other hand, the addition of Re and the formulation of Ru-Re bimetallic catalysts was favorable in all cases improving both conversion and selectivity to 1,2-PG. Early studies reported that the addition of a Re
2(CO)
10 precursor to the reaction medium markedly improved the activity and selectivity at 1,2-PG of the Ru/Al
2O
3 catalyst [
88]. Further studies showed that the formulation of Ru-Re catalysts supported on SiO
2, Al
2O
3, ZrO
2, TiO
2, and HZSM-5 and H-β type zeolites resulted in an improvement in activity levels, which was assigned to the dispersion of Ru particles by the addition of Re, suggesting a synergistic effect between the two species. The increase in selectivity to 1,2-PG was attributed to an inhibitory effect of Re on the formation of secondary products, such as 1-POH and 2-POH [
74]. The existence of ReOx particles interacting with Ru
0 particles has been found to be responsible for the enhancement of metal dispersion [
96]. On the other hand, such species impart higher acidity to the catalyst [
97]. Some reports have indicated that the enhancement in activity by Re also depends on the preparation method. Li et al. reported the preparation of Ru-Re bimetallic catalysts employing the deposition of a Ru-polyvinylpyrrolidone colloid followed by impregnation with the Re precursor. The results showed that, compared to the conventional impregnation method, this catalyst presented a better performance due to the smaller particle size and the high metal content that is possible to achieve with this preparation method [
98].
The addition of other metals, such as Co and Ni, had a positive impact on Ru activity levels. Feng et al. studied the addition of these metals to Ru/TiO
2 catalysts, showing that, although the conversion decreases, there is an increase in selectivity to 1,2-PG. The formulation of a Ru-Co catalyst proved to be the best, even using other supports such as SiO
2, ZrO
2, and Al
2O
3 [
100,
101,
102]. Li et al. added Fe to a Ru/CNT catalyst, promoting an improvement in the activity and stability of the catalyst due to the formation of iron oxides at the periphery of the Ru particles, in the form of FeO and FeO
1+x (0 < x < 0.5). These species interact synergistically with the Ru particles up to a Ru:Fe molar ratio = 2. The authors showed that the addition of higher amounts of Fe causes a drop in activity levels due to the surface blocking of Ru [
103].
The preparation of Ru-Cu bimetallic catalysts has also been reported in the literature. Liu et al. prepared catalysts supported on SiO
2, TiO
2, ZrO
2, Al
2O
3, and HY and NaY zeolites. Of all the prepared catalysts, Ru-Cu/ZrO
2 with a Cu:Ru molar ratio = 1:10 showed the best performance, due not only to the acidity of the support but also to the synergistic effect between Ru and Cu by electron transfer from Ru to Cu [
106]. Salazar et al. studied Ru-Cu/TiO
2 catalysts and found the maximum selectivity to 1.2-PG for a Cu:Ru mass ratio = 1:1, which was attributed to the interaction between Cu and Ru. The results indicated that the presence of Cu favors the formation of 1,2-PG by suppressing the formation of EG [
105].
With respect to obtaining 1,2-PG in vapor phase, the results indicate that Ru catalysts do not selectively promote the formation of 1,2-PG because thermal levels increase the ability for C-C bond cleavage. Ru/SBA-15 catalysts showed yields for 1,2-PG of 15%, even though the glycerol conversion was 75% at 260 °C, 0.1 MPa of H
2 and 2.21 h
−1 (WHSV) [
107]. Ru/γ-Al
2O
3 systems showed similar yields (17%) at 230 °C, 0.1 MPa of H
2 and 2.09 h
−1 (WHSV) [
108].
From the results above, it can be concluded that Ru-based catalysts are active in glycerol hydrogenolysis to 1,2-PG, with the conversion depending on the metal particle size and the acidity of the support. Favoring metal dispersion and increasing support acidity, high conversions can be obtained, although those properties should be optimized so as not to generate side products. As monometallic Ru-catalysts are not so selective to 1,2-PG, bimetallic formulations are required, with Ru-Co, Ru-Fe, and Ru-Cu being the most effective ones. From all the formulations, Ru-Cu/Bentonite resulted in being the most active (yield to 1,2-PG = 86.4%).
3.2. Pt Catalysts
With respect to Pt catalysts,
Table 3 summarizes the operating conditions and liquid-phase activity results employing batch reactors.
From the point of view of intrinsic metal activity, Pt catalysts have been demonstrated to be less active than Ru catalysts but have shown higher selectivity at 1,2-PG. Dasari et al. carried out the first studies on a screening of catalysts and determined that, comparatively, Pt/C is less active than Ru/C, but shows a higher selectivity to 1,2-PG [
45]. In this regard, Maris et al. determined that the higher selectivity at 1,2-PG is attributed to the lower C-C bond cleavage ability of Pt compared to Ru [
94].
With respect to other metal phases, such as Pd, Cu and Ni, Pt was found to be intrinsically more active. Von Held Soares et al. prepared Pt, Pd, and Ni catalysts supported on Fe
3O
4. The order of activity found was as follows: Pt > Pd > Ni. The results showed that, in the case of Pt, a spillover effect also contributes to improving the catalytic performance [
117]. Recently, Wei et al. prepared catalysts of Pt, Cu, Ru, and other bimetallic combinations supported on tungsten carbides (WC
x). The results showed that Pt/WC
x led to the highest yields for 1,2-PG [
115].
Of the Pt catalyst set, early studies employed commercial Pt catalysts [
45,
94,
112,
113], but the activity of catalysts prepared from H
2PtCl
6–6H
2O [
94,
110,
111,
114,
117,
119,
120,
121], Pt(NH
3)
4Cl
2H
2O [
109,
118], and (NH
4)
2PtCl
6 [
115] have also been evaluated. However, unlike Ru catalysts, papers have not reported studies on the particle size effect by the precursor type or preparation methods employed. Most of the studies have focused on the role of the support and its modification to achieve good metal dispersions and/or suitable surface acidic properties.
In this sense, the use of supports based on carbon ©, metal oxides such as MgO, Al2O3, SiO2-Al2O3, TiO2, ZnO, CeO2, and Fe3O4, zeolites of the H-ZSM5 and Hβ type and hydrotalcites (HTL) has been reported.
Within the group of carbonaceous supports, different types with varying specific surface areas (~250–1400 m
2 g
−1) have been employed. The Pt/C catalysts, prepared by vapor phase metal deposition on the supports, presented high metal dispersion with particle sizes between 1.3 nm and 2.5 nm, depending on the specific surface area of the support. These catalysts led to high levels of activity and stability [
116].
With respect to supports based on metal oxides, zeolites, and hydrotalcites, the results are diverse and varied. Yuan et al. studied Pt catalysts prepared on acidic and alkaline supports. Their results showed that the more alkaline supports allow high conversions and selectivity to 1,2-PG to be obtained. The order of activity found was as follows: HTL > MgO > Al
2O
3 > Hβ > H-ZSM5 [
109]. Other authors have found that acidic supports led to high levels of activity. Checa et al. studied Pt catalysts supported on Al
2O
3, CeO
2, La
2O
3, and ZnO. The order of activity according to the support used was as follows: La
2O
3 > CeO
2 > ZnO > Al
2O
3. The increase in activity was attributed to the increasing order of surface acidity of the supports [
111]. Gandarias et al. reported that an amorphous SiO
2-Al
2O
3 support allows the dehydration of glycerol to AcOH, due to its acidic properties [
112]. It has also been reported that the acidic sites present in TiO
2 supports are responsible for favoring dehydration reactions [
110].
These results indicate that both alkaline and acidic supports allow the preparation of selective Pt catalysts towards the formation of 1,2-PG. According to the dehydrogenation–dehydration–hydrogenation mechanism, the alkaline supports would favor the dehydration of glyceraldehyde (GLA) to 2-hydroxyacrolein (2-HA) while the acidic supports, on the other hand, would favor the dehydration of glycerol to acetol (AcOH) in the dehydrogenation–hydrogenation mechanism.
In order to increase the selectivity towards 1,2-PG production, some authors have reported the modification of supports with acidic promoters. Rodrigues et al. studied Pt catalysts supported on Al
2O
3 modified with Nb
2O
5. The presence of Nb oxides, which endows the support with Bronsted-type acidity, favors the reduction in the Pt particles and enhances the cleavage of the C-O bond of the glycerol molecule [
118].
Regarding the presence of acid sites in Pt catalysts, it has been reported that they should be as close as possible to the Pt atoms. In this regard, Du et al. modified a Pt/Al
2O
3 catalyst by the layered atomic deposition technique. Although there was no change in the total acidity of the catalyst, their results indicated that the acidic metal-site proximity enhances the bifunctional dehydration–hydrogenation mechanism [
114].
To decrease the ability for C-C bond cleavage and increase the selectivity to 1,2-PG, bimetallic Pt catalysts were studied employing Re [
122,
123], Ni [
120,
121], Au [
124], Sn [
125], Fe [
104], and Ir [
126].
Table 4 summarizes the operating conditions and activity results of liquid phase Pt bimetallic catalysts employing batch reactors.
The addition of Re to Pt/C and Pt/CNT catalysts was effective for the production of 1,2-PG due to the formation of a Pt-Re alloy with sizes smaller than 2 nm [
122,
123]. The addition of Fe to Pt/γ-Al
2O
3 catalysts also promoted higher activity towards the formation of 1,2-PG due to the generation of new active sites involving both Pt and Fe atoms [
104]. The Sn modification of Pt/SiO
2 resulted in the formation of Sn
+n species with Lewis-type acidity, upon which the cleavage of the C-O bond of the glycerol molecule is facilitated, allowing high selectivity to 1,2-PG [
125]. Recently, Liu et al. reported the modification of Pt/SiO
2 with Ir-ReO
x. The presence of ReO
x species favored C-O bond cleavage reactions, while Pt-Ir particles promoted H
2 formation for hydrogenolysis but, at the same time, were active for C-C bond cleavage reactions [
126].
The preparation of Pt-Ni catalysts on α-Al
2O
3-CeO
2-ZrO
2 [
120] γ-Al
2O
3 [
121] also allowed the performance of Pt catalysts to be improved due to the formation of a Pt-Ni alloy. For the case of Pt-Ni/α-Al
2O
3-CeO
2-ZrO
2, the species responsible for the increased activity was PtNi
3 [
120].
In contrast to Ru catalysts, the addition of Au to Pt catalysts improved the performance of the mono-metal catalysts. In this sense, bimetallic Pt-Au catalysts were prepared on MgO, SiO
2, sulphated ZrO
2, H-Mordenite, MCM-41, and TiO
2. Of all the catalysts studied, Pt-Au/TiO
2 was the most active one due to the higher dispersion of the Pt-Au particles (3.7 nm) [
124].
From the foregoing, Pt-based catalysts are more active intrinsically than Ru-based catalysts, but show higher selectivity to 1,2-PG. The election of the support plays a fundamental role, as its acidity or basicity favors the dispersion of the metal particles and enhances the catalytic activity. Alkaline supports have been demonstrated to be the most suitable for glycerol hydrogenolysis, being the most active Pt/HTL among all the catalytic formulations (yield to 1,2-PG = 85.6%).
3.3. Pd Catalysts
Pd-supported catalysts have been less explored in the literature than Ru and Pt, and their use has been limited to the production of 1,2-PG in the liquid phase.
Table 5 summarizes the operating conditions and activity results of liquid-phase Pd catalysts employing batch reactors.
Unlike Ru and Pt catalysts, Pd catalysts have been demonstrated to be more selective towards 1,2-PG formation due to their ability to cleave the C-O bond versus the C-C bond, although some were not very active. In this respect, Pd/SiO
2 [
127], Pd/C [
45], Pd/ZrO
2 [
87], and Pd/bentonite [
128] showed good selectivity to 1,2-PG (57–90%), but with low overall yields due to the low conversions obtained.
Most of the research work has focused on the appropriate preparation method to achieve high levels of conversion. Pd(NO
3)
2 [
127], PdCl
2 [
87,
119,
130,
131,
132], Pd(AcO)
2, and Na
2PdCl
4 [
129] have been used as precursors for the preparation of Pd catalysts, although the use of commercial catalysts has also been reported [
45,
127].
With respect to the precursor used, the following order of activity has recently been reported: PdCl
2 > Pd (NO
3)
2 ~ Pd(AcO)
2 for Pd/SBA-15 catalysts [
133]. It has been suggested that the presence of Cl
− from PdCl
2 improves the reducibility of the catalyst, generating a higher metal dispersion, and therefore improving the activity levels [
134].
Regarding the preparation, the co-precipitation method has been more efficient than the impregnation method, because it generates higher dispersions of the metal phase and therefore higher levels of activity [
130,
131]. In this sense, Pd/Fe
2O
3 [
131], PdO/CoO [
131], Pd/Co
3O
4 [
130], and Pd/Fe
3O
4 [
130] catalysts prepared by coprecipitation were more active than those prepared by impregnation.
The activation process has also been reported as a variable affecting activity. PdO/Fe
2O
3 [
131] and PdO/CoO [
131] catalysts showed 94% and 60% yields, respectively, under very similar reaction conditions, without being reduced in the H
2 flow. In the case of PdO/Fe
2O
3 [
132], its activity was higher than the activity of the reduced catalyst, Pd/Fe
2O
3 [
131]. It even showed higher activity than the Pd/Fe
2O
3 catalyst prepared by deposition–precipitation, whose support was obtained by hydrothermal treatment and tested under more severe reaction conditions [
129].
In order to improve activity levels, bimetallic Pd catalysts based on Pd-Re [
133,
135], Pd-Cu [
136,
137], Pd-Zn [
138,
139], and Pd-Ni [
140] have been used.
Table 6 summarizes the operating conditions and activity results of the bimetallic Pd catalysts in batch reactors.
Li et al. studied Pd-Re/SBA-15 catalysts and reported that the formation of ReOx particles increases the acidity of the catalyst and improves its reducibility, leading to improved activity levels [
133]. In another article, the authors reported that the addition of Re decreases the average Pd size, and the oxidation state of the ReOx particles is much higher in the Pd-Re/SBA-15 catalyst than in the Re/SBA-15 catalyst [
135].
Pd-Zn/ZrO
2 systems have also been demonstrated to be active and selective due to the oxophilicity of Zn which promotes the cleavage of the α-C-H bond in the 2,3-hydroxypropanoxide intermediate on the surface of the Pd-Zn alloy. The results showed that this is the determining step in the reaction rate and therefore the performance of the Pd-Zn catalyst is much better than Pd [
138]. The effect of Zn was also studied by Li et al., who found an improvement in activity by the formation of Pd-Zn nanoparticles [
139].
Pdx-Cu
0.
4/Mg
5.
6-xAl
2O
8.
6-x catalysts prepared by the co-precipitation method were found to be active and selective for the formation of 1,2-PG due to an enhanced ability to hydrogenate by a H
2 spillover process from Pd to Cu [
136]. The same effect was reported for Pd-CuCr
2O
4 catalysts. The presence of Pd allowed the reaction rate to increase about 1.7 times more than that obtained with CuCr
2O
4 catalysts. The H atoms stored in Pd diffused towards CuCr
2O
4 improving its reducibility and therefore increasing the number of surface-active sites formed from Cu
0 [
137].
According to the results, Pd-based catalysts are less active than Ru- and Pt-based catalysts, although they exhibit the highest selectivity to 1,2-PG due to the poor ability to cleave C-C bonds among the three noble metal phases. The preparation methods affect the catalytic performance of Pd-based catalysts due to the impact on metal dispersion. Although bimetallic formulations were implemented, the best results were obtained using PdO/Fe2O3 (yield to 1,2-PG = 94.0%).
3.4. Cu Catalysts
Within the group of catalysts based on non-noble metals (Cu, Ni, Co), the Cu catalysts, both supported and mass catalysts, have been the ones most widely used for the production of 1,2-PG, both in liquid and vapor phase.
Table 7 summarizes the operating conditions and activity results of the Cu bulk catalysts used in liquid phase in batch reactors.
Among the bulk catalysts, one of the first to be studied was Cu-Raney [
41,
127], obtained by leaching a Cu-Al alloy treated with NaOH. The reason for the high selectivity to 1,2-PG obtained with this catalyst (86%) is due to the intrinsic ability of Cu to cleave C-O bonds and the low ability to cleave C-C bonds, which decreases the selectivity to side products such as EG and gases [
41]. The modification of Cu-Raney with metal oxides such as MgO allows for the improved performance at 1,2-PG due to the formation of a dispersed Cu-MgO phase in the Cu-Raney pores [
149,
164].
CuCr
2O
4 catalysts and their variants were also active in the hydrogenolysis reaction. In the case of CuCr
2O
4, it has been reported that the yield to 1,2-PG is a function of the degree of catalyst reduction, necessary to achieve the formation of active Cu
0 and Cu
+ species in the Cr
2O
4 matrix [
45,
154]. The existence of a CuCr
2O
4 spinel favored the hydrogenation step, due to the ability to accumulate both H
2 and H bulk [
150]. Recently, a theoretical study with DFT-based calculations revealed that the formation of AcOH is both thermodynamically and kinetically favored on the surface of the Cu and CuCr
2O
4 spinel. The spinel structure provides suitable sites for the adsorption of glycerol, its dehydration to AcOH, and subsequent hydrogenation to form 1,2-PG [
165]. Similarly, it has been reported that another CuFe
2O
4-type spinel, present in Cu-Fe catalysts, was also active for the formation of 1,2-PG [
145].
Other variants of the CuCr
2O
4 catalyst have been evaluated, which improved the yield to 1,2-PG under certain reaction conditions. The incorporation of Ba, for example, allowed stabilization and avoided the sintering of the Cu
0 particles through the formation of BaCrO
4 [
142]. The preparation of a Cu-Cr system, through the sol–gel technique in the presence of propylene oxide, allowed an active catalyst to be obtained due to the presence of CuCr
2O
4, CuO, and Cr
2O
3 phases whose percentage content in the catalyst depends on the Cu:Cr molar ratio [
146,
166].
On the other hand, Cu-ZrO
2 systems prepared by coprecipitation showed low yields for 1,2-PG (9%) [
141]. Their modification with MgO and the optimization of the Cu:ZrO
2 ratio allowed the yield to be improved to 1,2-PG (59%), due to the basic properties of MgO [
109]. In fact, for Cu/MgO catalysts the activity was found to be a function of the basicity of the support, in addition to the dispersion of Cu and MgO particles formed during the catalyst preparation [
153,
167]. Recently, it has been reported that a Cu-MgO catalyst, with a Cu:MgO molar ratio = 0.5, led to 1,2-PG yields of 37% at 200 °C in the absence of H
2 pressure, due to in situ H
2 generation by glycerol reforming [
168].
The replacement of MgO by SiO
2 and ZnO were also tested alternatives in the preparation of Cu catalysts. CuO/SiO
2 catalysts prepared by co-precipitation with colloidal SiO
2 showed acceptable yields at 1,2-PG (69%) due to the presence of highly dispersed Cu
0 particles, and Cu
+ particles that enhanced the metal–support interaction, avoiding sintering [
151,
169].
With respect to the Cu-ZnO catalysts, the activity was found to be dependent on the Cu:Zn molar ratio, the preparation method, and the Cu
0 particle size. The Cu:Zn molar ratio determines the formation of Cu and ZnO phases. In this regard, it has been reported that the formation of Cu and Zn hydrocarbonates with different crystalline phases, such as single-phase aurichalcite, favors the formation of highly interacting Cu with ZnO [
152]. The coprecipitation method has been employed in the presence of urea [
48,
62] and oxalic acid [
144] as precipitation agents, the latter being the best alternative due to the formation of CuO with higher surface area. The reduction in CuO generates small Cu
0 particles that improve the activity and selectivity to 1,2-PG in both the urea [
70] and oxalic acid [
144] coprecipitation methods. Gao et al. used the previously reduced Cu-ZnO catalyst in a continuous flow reactor at 250 °C, 2 MPa of H
2, and 7.6 h
−1 (WHSV). The catalyst was stable for 200 h of reaction, with conversions of 93–96% and selectivity to 1,2-PG of 79–83% using crude glycerol and analytical grade glycerol, respectively. For a Cu:Zn molar ratio = 1.86, the maximum yield to 1,2-PG is achieved due to the maximum exposed Cu surface area [
170].
More complex catalytic systems based on Cu, Zn and Al [
127,
156,
160,
171,
172,
173,
174], Cu, Zn and Ga [
159], Cu, Zn, Mg and Al [
77,
81,
87,
88] and Cu, Zn, Cr and Zr [
162] showed higher yields for 1,2-PG than Cu/ZnO.
In Cu, Zn, and Al catalysts, the presence of Al
2O
3 provides a higher number of acid sites that favor the formation of 1,2-PG and modify the textural properties. In this sense, an improvement in the selectivity to 1,2-PG has been observed due to a higher macropore fraction (from 1 to 10 μm) by the presence of Al
2O
3 [
156]. On the other hand, the preparation method has a direct impact on activity due to the type of species formed from both Cu [
101] and metal oxides [
173]. Comparing the traditional coprecipitation method in the presence of KOH and K
2CO
3 with coprecipitation via oxalate gel formation and mechanical mixing of oxides, the former allows higher yields to be achieved at 1,2-PG [
174].
In catalysts based on Cu, Zn, Mg, and Al, on the other hand, the activity is a function of the basic properties of the support and the dispersion of the Cu metal particles [
104,
105]. It has been shown that the presence of ZnO generates a strong interaction with Cu, allowing the formation of small Cu metal particles with a high dispersion and good reducibility, which allows high yields to be reached for 1,2-PG [
161]. In a liquid-phase continuous flow reactor, Zhou et al. employed a Cu-ZnO-MgO-Al
2O
3 catalyst with a Cu:Zn:Al molar ratio of 1:1:0.5, obtaining a glycerol conversion of 81% with 93% of selectivity to 1,2-PG at 200 °C, 4 MPa of H
2 and 4.6 h
−1 (LHSV) [
175].
Other catalysts, based on Cu and Al, were also studied in the hydrogenolysis reaction. Indeed, it has been demonstrated that these catalysts showed a better performance than CuCr
2O
4 catalysts [
154]. In these systems, the activity is a function of the Cu:Al molar ratio [
147] and the preparation method. In all cases, the co-precipitation method was used, varying the precipitating agent. The results showed that the highest yields are obtained using NaOH as precipitating agent [
176]. On the other hand, it has been reported that the presence of Cu
0 and Cu
+ species in the form of Cu
2O improves the performance of the catalysts [
176,
177], in addition to others that improve the activity levels such as CuAl
2O
4 [
177], CuAlO
2, and CuAl
4O
7 [
154]. In particular, the CuAl
2O
4 phase improves the reducibility and the ability of Cu to readily adsorb and desorb H
2 [
158].
Modification of Cu and Al catalysts with Ca led to an improvement in the acid-base properties of the catalyst by increasing the number of basic sites and allowing better dispersion of the Cu metal particles, leading to higher activity levels [
143,
155]. Mizugaki et al. prepared catalysts based on Cu nanoparticles in an amorphous aluminum oxide matrix (Cu-Al
2Ox) from the reduction in Cu hydrotalcites. Their results showed very high yields for 1,2-PG: they obtained a glycerol conversion of 100% with a selectivity to 1,2-PG of 98% using very dilute solutions of glycerol in 1,4-dioxane [
163].
Table 8 and
Table 9 summarize the operating conditions and activity results of liquid-phase Cu-supported catalysts employing batch reactors and continuous flow reactors, respectively.
As with other metal phases, it has been reported that both the acidity of the support and the particle size are two important properties to take into account in catalyst design.
Most of the Cu-based supported catalysts have been prepared by the incipient wet impregnation technique, using Cu(NO
3)
2 as a precursor [
127,
151,
152,
181,
189,
191,
193,
194,
195,
196,
197,
198,
199,
200,
201]. However, other methods such as co-precipitation [
151,
182,
183,
184], deposition–precipitation [
185,
202,
203,
204], and ion exchange [
190,
197] have been reported.
In the study of Cu catalysts, different supports have been reported, such as γ-Al
2O
3 [
185,
187,
189,
191,
195,
199,
205], SiO
2 [
151,
186,
192,
202,
204], ZnO [
182], TiO
2 [
178,
199], MgO [
179,
185] and Cr
2O
3 [
127], dealuminated ultra-stable Y-type zeolites (DUSY) [
183] and HZSM-5 [
188], mesoporous silica (HSM) [
180], SBA-15 [
198], and hydrotalcites [
184,
188].
For acidic supports, such as ZnO [
182], Cr
2O
3 [
127], γ-Al
2O
3 [
185,
187,
189,
191,
195,
199,
205], and DUSY zeolite [
183], it has been reported that the acid sites of the support favor the dehydration of the glycerol to AcOH which then hydrogenates over the Cu metal sites; however, the Cu active phase also generates the C-O bond cleavage.
On the other hand, when the acidity of the support is high, as in the case of zeolite HZSM-5 [
188], the selectivity to 1,2-PG decreases due to the dehydration of glycerol to acrolein. Other supports such as TiO
2 have acidic sites that favor the formation of side products such as 1-POH [
178,
199].
One of the most widely studied catalysts has been Cu/γ-Al
2O
3 [
178,
185,
187,
195,
199]. In these systems, the selectivity to 1,2-PG has always been high (~90%) regardless of the Cu content of the catalyst [
185]. It has been reported that the method of preparation of the Cu/γ-Al
2O
3 catalyst affects the catalytic performance. Vila et al. studied the preparation of these catalysts and their results indicated that the catalytic performance strongly depends on the calcination, reduction, and oxidation processes. In these processes, the presence of Cu
0, Cu
+, and Cu
+2 species was evidenced. The results showed that the activity and selectivity to 1,2-PG increases in the presence of Cu
0 and Cu
+ species, while Cu
+2 species are the least active in the hydrogenolysis reaction [
195].
Regarding the γ-Al
2O
3 sites, a DFT study revealed that the hydrogenolysis of glycerol is facilitated by the presence of the acidic sites found on the alumina surface. The hydroxylation of these sites in the presence of water generates similar activity in terms of glycerol adsorption and AcOH formation as the Cu metal sites. However, the activation energy is lower in the case of the alumina surface sites, allowing high conversions to be achieved using this support [
205].
The best results with Cu/γ-Al
2O
3 were obtained in continuous flow reactors with 1,2-PG yields of 70% at 250 °C, 4 MPa of H
2, and 2 h
−1 (WHSV) [
187].
The modification of γ-Al
2O
3 with promoters has also been a variable studied in the literature. Heteropoly acids of the H
2SiW
12O
40 type have allowed the reducibility of Cu particles to be improved on the support and to increase the acidity of the catalyst without promoting side reactions [
189]. The modification with H
3BO
3 was also beneficial due to an effect on the dispersion of the Cu particles and the improvement of their hydrogenating capacity [
191].
For neutral supports, such as SiO
2, SBA-15, and other mesoporous silica (HSM), the published articles focused on the metal particle size. With respect to Cu/SiO
2 catalysts, the preparation of the catalysts by the impregnation and co-precipitation methods led to better activity results with respect to the bulk Cu/SiO
2 catalysts. Of the two methods, the co-precipitation method led to a greater dispersion of the Cu particles and better metal–support interaction [
151,
202,
204]. Other more sophisticated preparation methods have shown even better performances for the Cu/SiO
2 catalyst. For example, preparation by the hydrothermal ammonia evaporation method led to Cu
0 species in the form of nanoparticles dispersed on the support surface. In these catalysts, the cooperative effect of Cu
0 and Cu
+ species are indicated to be responsible for the catalytic performance [
192]. Another preparation, by coating SiO
2 with Cu from a Cu-polyvinylpyrrolidone colloid, led to high yields for 1,2-PG [
186].
The use of mesoporous silicas, such as SBA-15 and hexagonal mesoporous silica (HSM), allowed more active catalysts to be obtained than Cu/SiO
2 [
180], due to their textural properties of mesoporosity and specific surface area, which allowed high dispersions to be achieved in the active phase [
190,
198].
For basic supports, such as MgO [
179,
185] and hydrotalcites [
184,
188], the literature reports have indicated that higher basicity increases the levels of glycerol conversion and selectivity to 1,2-PG. It has also been reported that both MgO and hydrotalcites help to disperse Cu particles, which contributes to an improvement of the catalyst performance [
179,
184].
Since Cu is an active phase with good C-O bond cleavage ability, the incorporation of another metal in the catalyst design aims to increase the conversion of glycerol with respect to monometallic catalysts. In this regard, the study of Cu-Ru [
206], Cu-Ag [
196,
207], Cu-Ni [
208,
209], and Cu-Zn [
203,
210] catalysts has been reported.
Table 10 summarizes the operating conditions and activity results of bimetallic Cu catalysts in batch reactors.
In Cu-Ag/γ-Al
2O
3 catalysts, it has been reported that the addition of Ag generates a greater dispersion of Cu particles and facilitates their reduction, generating Cu
+ species that are active in the hydrogenolysis of glycerol [
196,
203].
The study of Cu-Ni catalysts was reported on γ-Al
2O
3. Employing Cu-Ni/γ-Al
2O
3 with a Cu:Ni mass ratio = 3, Pudi et al. reported a 1,2-PG yield of 26% at 250 °C, 1 MPa of H
2, and 5 h of reaction, assigning the result to the formation of a Cu
0.
75Ni
0.
25Al
2O
4-type mixed oxide [
208]. Similar results with yields for 1,2-PG of 24% were obtained with a Cu-Ni/γ-Al
2O
3 catalyst in a continuous flow reactor at 250 °C, 4 MPa of N
2, and 2 h
−1 [
187]. In both cases, the catalyst was prepared by wetness incipient impregnation. Yun et al. prepared the Cu-Ni/γ-Al
2O
3 catalyst by coprecipitation using mesoporous alumina, which leads to a better dispersion of the metal phase. For a Cu:Ni molar ratio = 9:1, the catalyst showed the best activity, and it became evident that the increase in the surface ratios of Ni
0/(Ni
0 + Cu
0) and Cu
0/(Cu
0 + Cu
+2) was responsible for the increase in yield to 1,2-PG [
209]. The highest yield obtained with Cu-Ni/γ-Al
2O
3 systems was 57.8% at 220 °C, 4 MPa of H
2, and 24 h [
209].
The incorporation of Ru to Cu catalysts supported on carbon nanotubes (CNT) resulted in an increase in activity towards the formation of 1,2-PG, because it favored the dispersion of Cu particles and promoted their hydrogenation capacity by a reactive H spillover process from Ru to Cu [
206]. The same spillover effect was found in Cu-Zn catalysts supported on MgO [
210]. In these systems, the presence of Zn enhances the reducibility of CuO species and favors the dispersion of Cu
0 particles, while providing suitable acid sites to improve the glycerol dehydration step [
203].
Table 11 summarizes the main catalysts evaluated in vapor-phase continuous flow reactors.
From the set of Cu bulk catalysts studied, Cu/ZnO/ZrO
2 and Cu/ZnO/TiO
2 showed the lowest yields for 1,2-PG (12 and 15%, respectively). Although the catalysts showed high selectivity at AcOH (60–76%), due to the presence of TiO
2 and ZrO
2, the low selectivity to 1,2-PG could be due to the low H
2 pressure (0.1 MPa) employed in the vapor phase condition [
211].
Cu/Cr
2O
3 [
213] and Cu/ZnO/Al
2O
3 [
127] bulk catalysts, however, allowed yields to be obtained for 1,2-PG of 77% and 88%, respectively, even under less severe reaction conditions. In these catalysts, the acidic properties of the supports with the ability to dehydrate the glycerol to AcOH are combined with the presence of metallic Cu with the ability to hydrogenate the AcOH produced during the reaction.
The most studied Cu-supported catalysts have been the Cu/γ-Al
2O
3 ones [
193,
201,
217,
219]. In these systems, the activity levels depend on both the presence of Cu metal particles [
217] and their exposed metal surface [
201]. Sato et al. evaluated Cu/γ-Al
2O
3 catalysts in vapor-phase discriminating between dehydration and hydrogenation reactions. The authors indicated that the presence of Cu not only allows the hydrogenation of AcOH to 1,2-PG in H
2 flow, but also intervenes in the dehydration of glycerol to AcOH when the reaction is carried out in N
2 flow. At 250 °C, 0.1 MPa of N
2 and 7.6 h
−1 (WHSV), the authors reported a yield to 1,2-PG of 50% [
193]. Optimizing the operating conditions and Cu content (15 wt.%), Dieuzeide et al. reported yields of 60% at 200 °C, 0.1 MPa of H
2 and 30.6 h
−1 (WHSV) [
219]. Of all the studies reviewed to date, the maximum yield to 1,2-PG obtained with Cu/γ-Al
2O
3 was 96.8% at 230 °C, 1.4 MPa of H
2 and 2.9 h
−1 (WHSV) [
217].
The modification of Cu/γ-Al
2O
3 catalysts with Ni and Ag allowed for an increase in the activity levels. In Cu-Ni/γ-Al
2O
3 catalysts, the presence of a Cu-Ni bimetallic site with a high exposed surface area and dispersion was responsible for the high yield obtained for 1,2-PG [
214,
220], while the presence of Ag in Cu-Ag/γ-Al
2O
3 catalysts allowed inhibiting undesired reactions that lead to the formation of side products, such as EG, improving the selectivity for 1,2-PG [
218].
For other supported catalysts, dispersion and surface area effects, as well as acidic properties, have also led to high yields for 1,2-PG. Bienholz et al. used Cu/SiO
2 catalysts and determined that the rate of the dehydration and hydrogenation reactions depends linearly on the exposed Cu metal surface [
197]. In other catalytic systems, based on basic supports such as MgO [
216] or neutral ones such as SBA-15 [
200], it has been reported that Cu dispersion is fundamental to reach high levels of activity. In the case of acidic supports, such as zeolites, both the high dispersions of the Cu metal phase and the acidic properties of the support favor the formation of 1,2-PG [
212].
Recently, Pandey et al. tested Cu-Zn/MgO catalysts in the vapor phase, finding that the presence of Cu
+2 and Cu
+ species facilitate the dehydration of glycerol to AcOH, while Cu metal particles hydrogenate AcOH to form 1,2-PG [
214].
From the results obtained, it can be concluded that Cu-based catalysts exhibit high selectivity to 1,2-PG due to their ability to cleave C-O bonds. In the presence of neutral supports, high metal dispersion led to an increase in catalytic activity, with the species Cu0 and Cu+ being the most active. On acidic supports, Cu activity is enhanced, but other secondary products can be formed. The presence of other metals forming bimetallic phases enhance glycerol conversion at the expense of a decrease in 1,2-PG selectivity. The best results were obtained with Cu-Al2Ox (yield to 1,2-PG = 99.0%)
3.5. Ni Catalysts
In contrast to Cu catalysts, Ni catalysts (both bulk and supported) have not been so widely studied in the literature and have been used to obtain 1,2-PG in the liquid phase, employing batch reactors. The catalytic results of the Ni bulk catalysts are presented in
Table 12.
Ni/Mg/Al catalysts, prepared by the coprecipitation method, showed low yields for 1,2-PG (2–3%), even when modified with Co as promoter [
172]. Similar yields (5%) were obtained with a Ni
3P bulk catalyst synthesized by a hydrothermal method at 150 °C in the presence of an ammonia solution of NiH
2PO
2 and NH
4H
2PO
2 [
221]. In the latter case, although the selectivity to 1,2-PG was 86%, the low yield was due to the low conversion achieved (5%), which could be associated with the reaction temperature (190 °C) and the high m
gly/m
c ratio (100).
Higher yields for 1,2-PG (18%) were obtained with a commercial Ni-Raney catalyst at 230 °C, 4 MPa of H
2, and 9 h reaction time [
222]. At higher reaction times it was possible to obtain yields of 26% (24 h) [
45] and 69% (44 h) [
51]. For this catalyst, higher reaction temperatures promoted C-C bond cleavage reactions leading to the formation of side products [
222]. The addition of Ag to Ni-Raney led to the formation of new Ni-Ag metal sites that suppressed C-C bond cleavage reactions [
223]. A similar effect was found in ZnNiAl-type hydrotalcites, which were also found to be active and selective to 1,2-PG formation. In these systems, the formation of a Ni-Zn alloy inhibits C-C bond cleavage reactions and enhances the adsorption of the terminal hydroxyl group of glycerol by promoting C-O bond cleavage [
224].
Table 13 summarizes the operating conditions and activity results of Ni-supported catalysts in batch reactors.
Supported catalysts based on Ni as the active phase showed, in general, high selectivity to 1,2-PG, comparable to those obtained with supported catalysts based on Ru and other noble metals. Although their ability to hydrogenate was lower than that of the noble metal-based catalysts, this was compensated for by the use of higher reaction temperatures and pressures, which led, on the other hand, to a higher deactivation by sintering of the active phase [
232].
With respect to other metal phases based on non-noble metals, commercial Ni/C catalysts were demonstrated to be as active and equally selective to 1,2-PG as Cu catalysts [
45,
227]. The same trend was observed for commercial Ni/SiO
2-Al
2O
3 catalysts compared to CuCr
2O
4 catalysts; however, the selectivity towards C-C bond cleavage was higher for the Ni catalyst [
45].
Some authors have reported the study of commercial catalysts such as Ni/SiO
2-Al
2O
3, where good particle dispersion was observed, so the hydrogenolysis reaction was carried out in a N
2 atmosphere and the H
2 was generated via the aqueous phase reforming of glycerol (APR) [
233,
234]. Maximum yields for 1,2-PG of ~29% were obtained at 240 °C, 3.3 MPa of N
2, and 4 h [
233]. The same result was obtained at 200 °C, 2.5 Mpa of H
2, and 8 h of reaction [
227].
The articles that have prepared and studied Ni-supported catalysts have mainly used Ni(NO
3)
2 [
127,
181,
225,
226,
229,
231,
233,
234,
235,
236,
237,
238] as the precursor, the impregnation method being the most widely used technique, although the co-precipitation [
235] and deposition–precipitation methods have also been reported [
226,
237]. The most commonly used supports are based on activated carbon (AC) [
229,
239], SiO
2 [
191,
225,
236], γ-Al
2O
3 [
127,
181,
225,
230,
240], CeO
2 [
226,
237] and WO
3 [
231], and zeolites of the NaX, NaA, NaZSM-5, and NaMOR types [
225].
Ni is a metal capable of participating in both C-O and C-C bond cleavage reactions due to its high electron density. In the presence of inert supports, the formation of products depends exclusively on the structural characteristics of the Ni metal phase. An example of catalytic systems where this phenomenon occurs is Ni/SiO
2 [
127,
225,
236]. Ni/SiO
2 catalysts showed low activity (8–57%) with a low selectivity to 1,2-PG (44–60%) under moderate reaction conditions at 200 °C, 5–6 Mpa of H
2, and 10 h [
127,
225]. The results obtained could be assigned to the ability of Ni to participate in C-C bond cleavage reactions in the presence of a neutral support [
236].
In the presence of acid supports, different results are observed. For some catalysts based on acidic supports, such as Ni/γ-Al
2O
3 and Ni/CeO
2, a certain tendency towards the formation of side products, such as EtOH, EG, 1-POH, and MeOH, was observed [
127,
225,
226,
230].
The modification of acidic supports with different basic promoters allows for a reduction in the formation of side products. When Ni/CeO
2 was modified with MgO, a basic species, the selectivity to 1,2-PG was improved, probably due to a balancing effect of the acid-base properties by addition of MgO [
237]. A similar effect was found for Ni/NaX catalysts, where the Na
+ content allows the acidity of the catalyst to be moderated [
225].
In other acid-supported Ni catalysts, however, the formation of side products was inhibited by the intrinsic characteristics of the acidic species. Ni catalysts supported on a modified sepiolite (MSEP) with WO
3, for example, showed yields for 1,2-PG of 81% due to the high glycerol conversions achieved and a high level of selectivity for 1,2-PG. The results were attributed to the presence of WO
3, with acidic characteristics, which allows the proper dispersion of Ni particles and inhibits C-C bond cleavage reactions [
231]. In another work, the acidic properties of Ni catalysts supported on a silica–carbon composite (Ni/SiO
2-C) were investigated, and it was determined that the presence of the carboxylic groups of the support promotes the dehydration of the glycerol producing AcOH, which then leads to the formation of 1,2-PG [
228].
In order to improve the activity of Ni catalysts, the preparation of bimetallic catalysts has also been studied in the literature by several authors. The results of liquid phase activity employing batch reactors are shown in
Table 14.
The use of bimetallic Ni catalysts with Ce [
239,
243], Ir [
241], Cu [
181,
235,
240], Zn [
242], and P [
244] has been reported.
Ni-Ce/SBA-15 catalysts showed better performances than Ni/SBA-15 catalysts due to a better dispersion of Ni on the support and a higher acidity provided by Ce. However, the maximum yields for 1,2-PG were 15% due to the low selectivity levels obtained [
239]. In Ni-Ce/AC systems, the addition of Ce led to higher levels of selectivity to 1,2-PG, with yields of 59%. In this case, the same effect on Ni dispersion was reported due to the presence of Ce in the form of CeO
2 [
243].
In order to improve the selectivity of Ni catalysts, Ir was used as a promoter for the formulation of Ni-Ir/γ-Al
2O
3 catalysts. Although the presence of Ir
+δ species (0 < δ < 4) allowed selectivity values to be obtained to 1,2-PG of 83%; the yields achieved were low (20%) due to the low conversions obtained [
241].
Ni-Cu/γ-Al
2O
3 catalysts prepared using the incipient wetting impregnation method [
181] and the sol-gel method [
235,
240] showed maximum yields for 1,2-PG of 67% and 74%, respectively. In the first case, an adjustment of the Cu:Ni mass ratio (1:1) allowed the maximum level of activity to be obtained without losing selectivity to 1,2-PG [
181]. In the second case, using a Cu:Ni ratio of 0.72:1, the maximum yield for 1,2-PG was obtained in the presence of formic acid as the H
2 donor substance [
235]. In both cases, the results were attributed to the formation of a highly dispersed Ni-Cu alloy [
181] that promoted C-O bond cleavage and enhanced the activity with respect to Ni/γ-Al
2O
3 and Cu/γ-Al
2O
3 catalysts [
235]. However, it was observed that the deactivation of the bimetallic catalyst was more pronounced due to higher coke generation and coke deposition on the active sites [
240].
For other catalytic systems such as Ni-Zn/SiO
2-C, the incorporation of Zn by means of the organometallic technique of surfaces on metals in the range 1.1–1.8 wt.% was responsible for the increase in activity and selectivity to 1,2-PG, due to the formation of an α-NiZn alloy that promotes the cleavage of the C-O bond in the glycerol molecule [
242].
In Ni-P/γ-Al
2O
3 catalysts, on the other hand, the presence of P decreases the degree of Ni reducibility, generating NiO species that increase the selectivity to 1,2-PG, but decrease the glycerol conversion. The overall yield for 1,2-PG increases due to the presence of -POx groups generated by the presence of P [
244].
With respect to obtaining 1,2-PG in the vapor phase, not many studies have employed Ni catalysts, due to the ability of this metal phase to form side products such as EG, EtOH, and gases. Ni/ZnO catalysts prepared by the quenching method on carbon spheres showed 1,2-PG yields of 45% at 235 °C, 3.1 MPa of Ar and 0.8 h
−1 (WHSV). The results were attributed to the high dispersion of Ni particles on the support and space velocity optimization [
245]. On the other hand, Ni-Ag/γ-Al
2O
3 systems showed similar yields for 1,2-PG at 200 °C, 0.1 Mpa, and 2.01 h
−1 (WHSV), which were attributed to the higher dispersion of Ni due to the presence of Ag [
238].
From the results above, it can be concluded that Ni-based catalysts are less active than Cu-based catalysts, showing lower selectivity values to 1,2-PG due to a higher ability to cleave C-C bonds. Better catalytic results were obtained in the liquid phase due to the generation of side products in the vapor phase. In order to increase catalytic activity, bimetallic formulations have been performed, although the best results were obtained modifying the acidity of the support, Ni/WO3-MSEP (yield to 1,2-PG = 85.4%).


 ,
, 























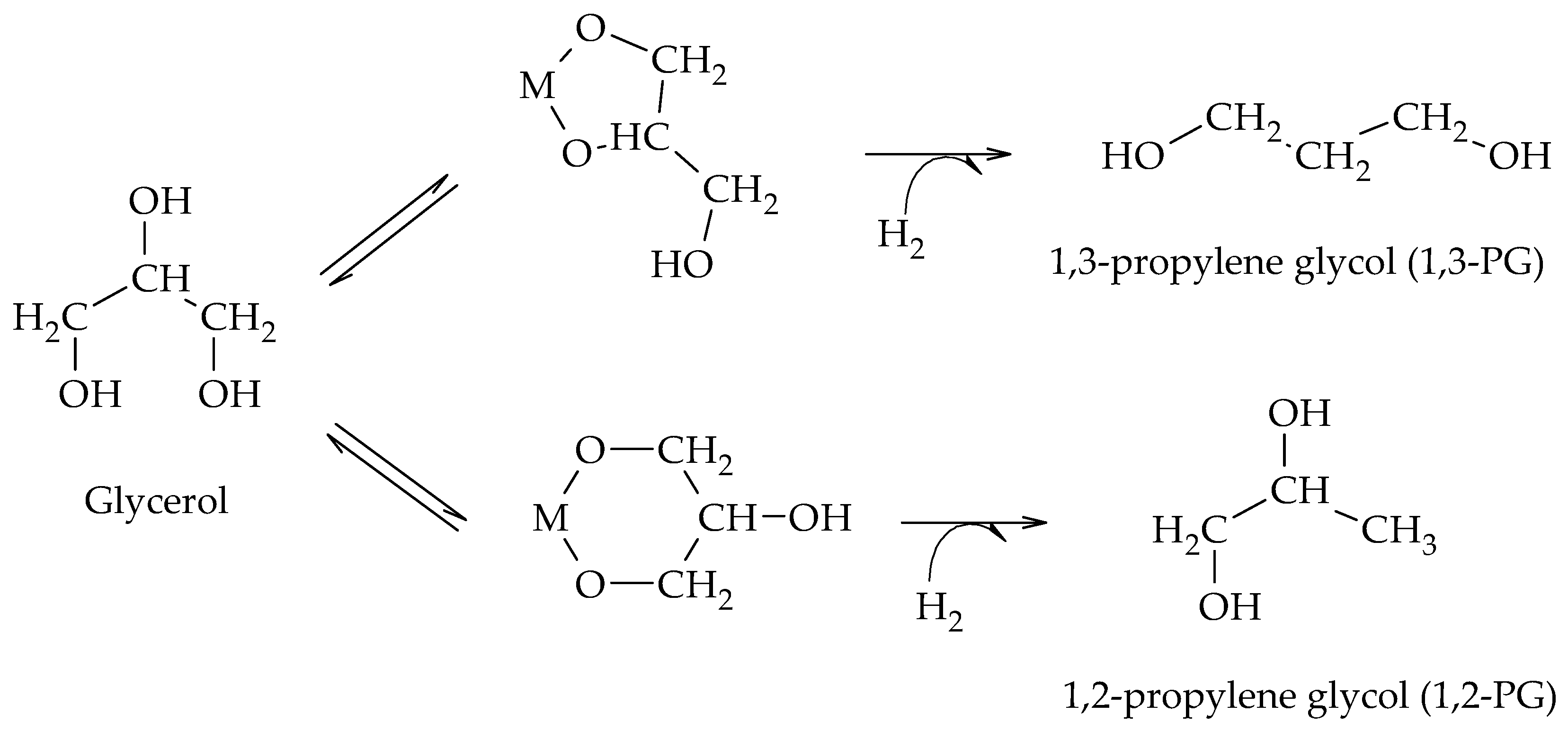





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}