
Preparation and Characterization of Calcium Carbonate Masterbatch–Alkali Soluble Polyester/Polyester Porous Fiber via Melt Spinning

Abstract
:1. Introduction
2. Experiment
2.1. Materials and Instruments
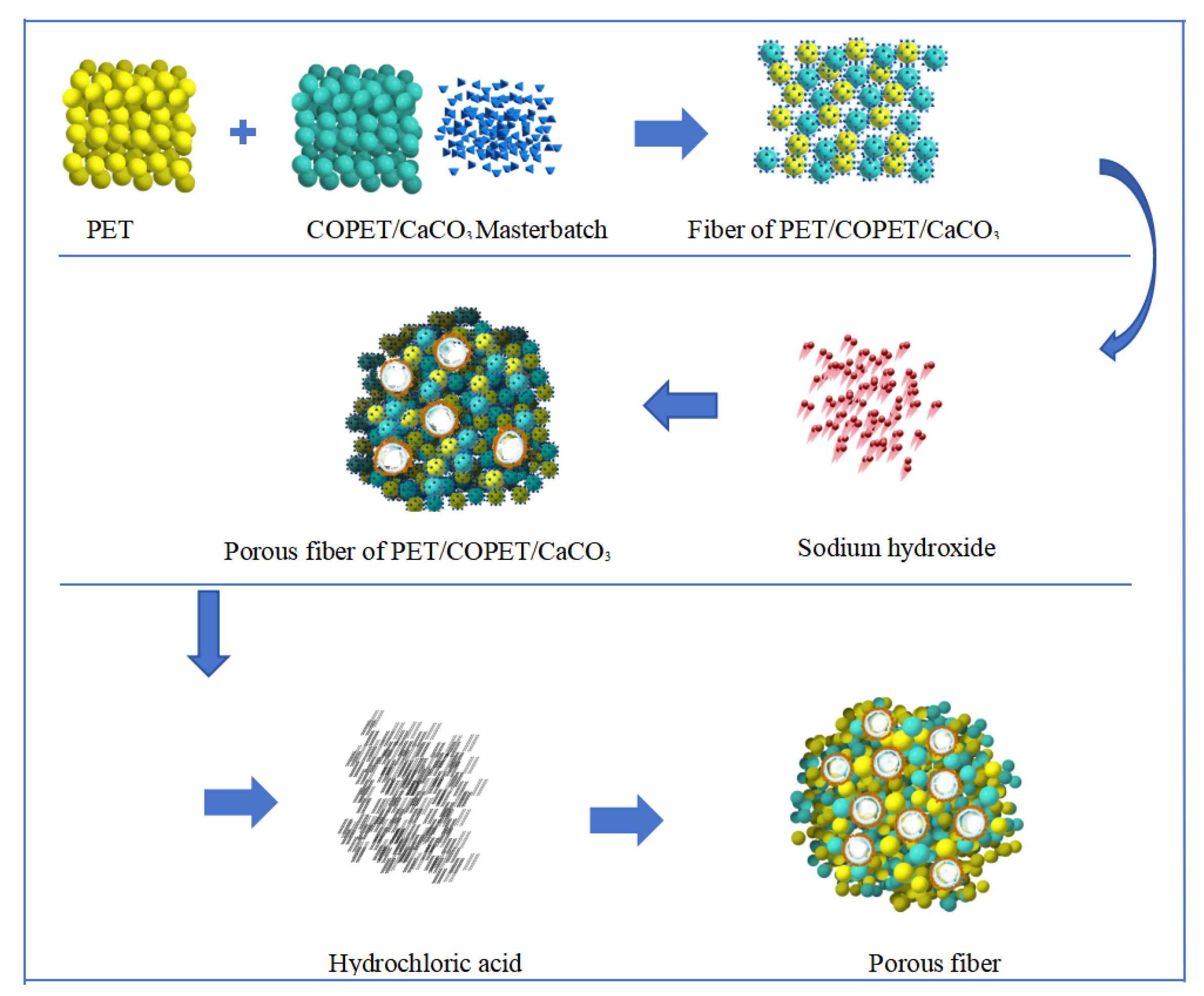
2.2. Preparation of Porous Fibers
2.2.1. Melt Spinning
2.2.2. Postprocessing
2.3. Characterization
2.3.1. Base (Acid) Decrement
- ①
- Alkali treatment alkali decrement
- ②
- Acid treatment acid decrement
2.3.2. Strength and Linear Density
2.3.3. Tungsten Filament Scanning Electron Microscope
2.3.4. Specific Surface Area and Aperture Distribution
2.3.5. DSC and TG
2.3.6. Fourier Infrared Spectrum
2.3.7. X-ray Diffraction
3. Results and Discussion
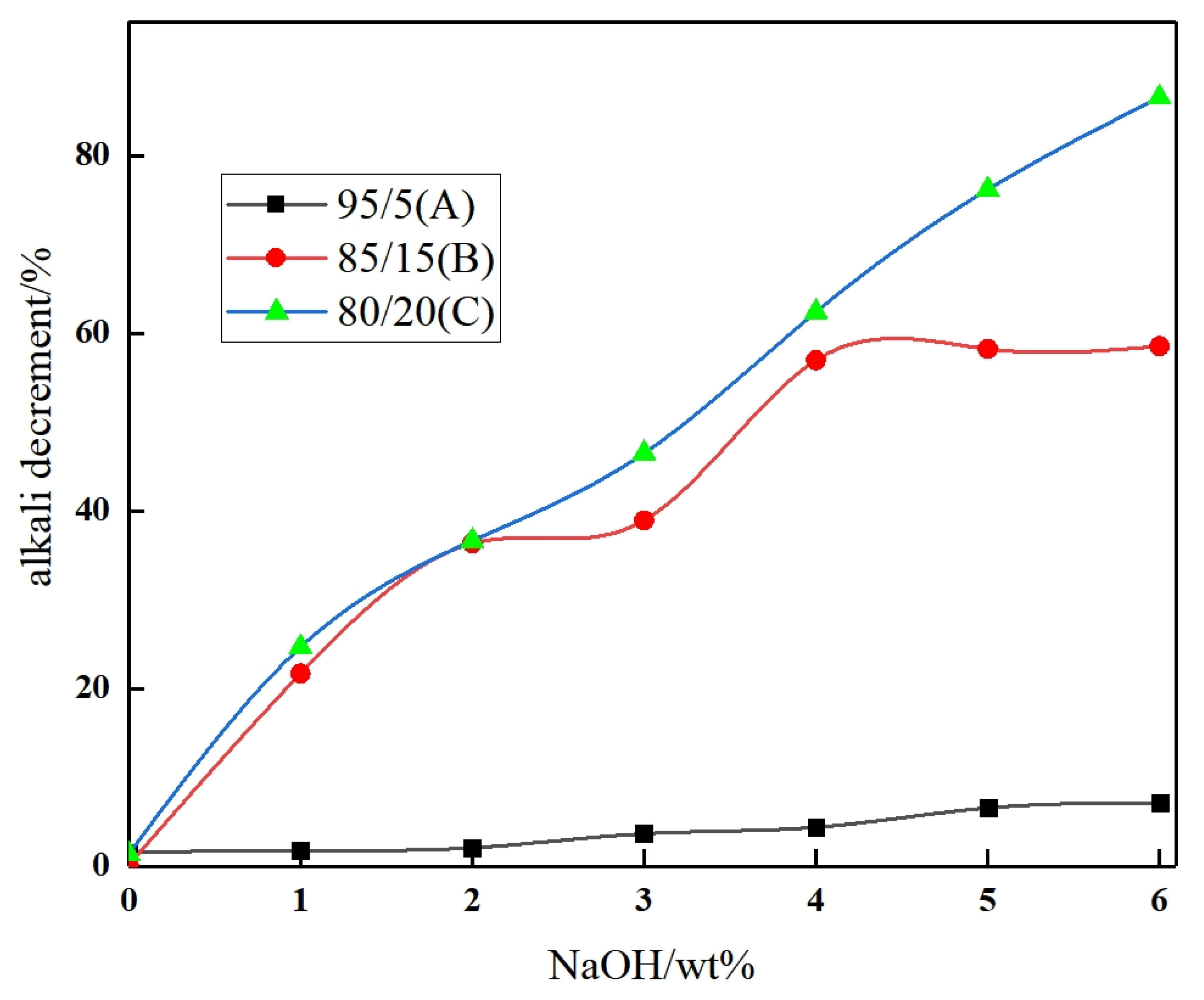
3.1. Base (Acid) Decrement
3.1.1. Alkali Decrement
3.1.2. Acid Decrement
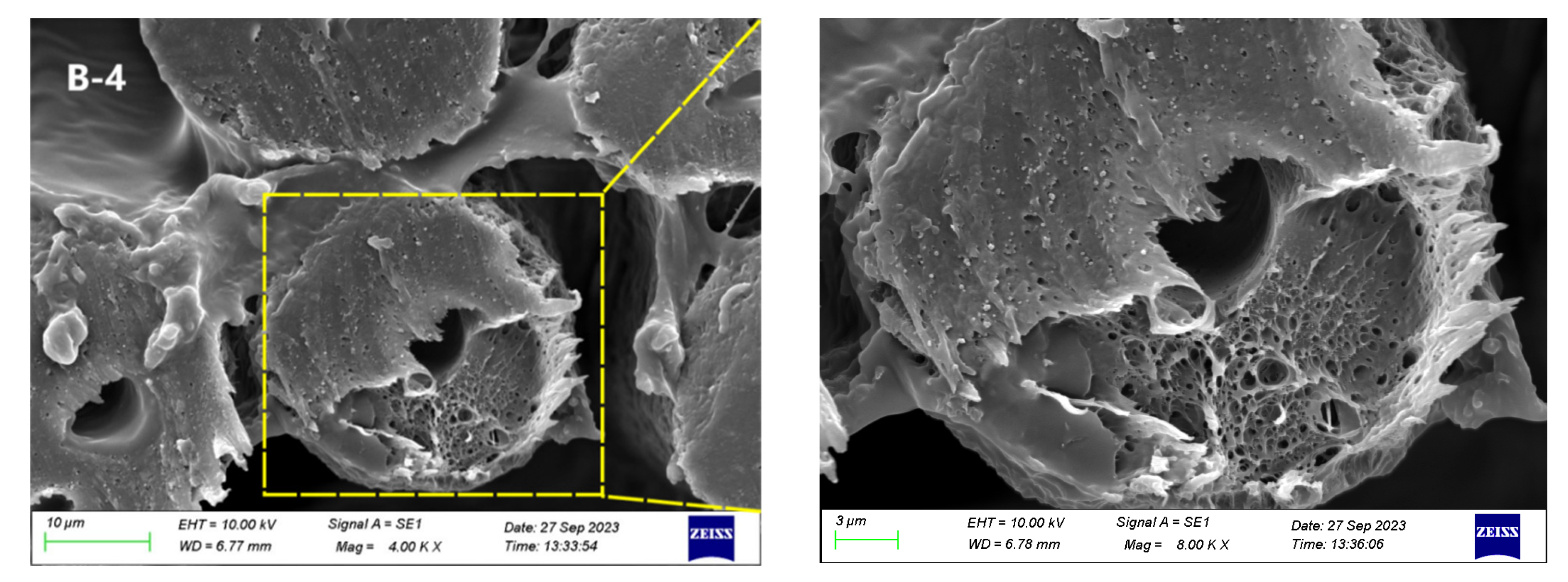
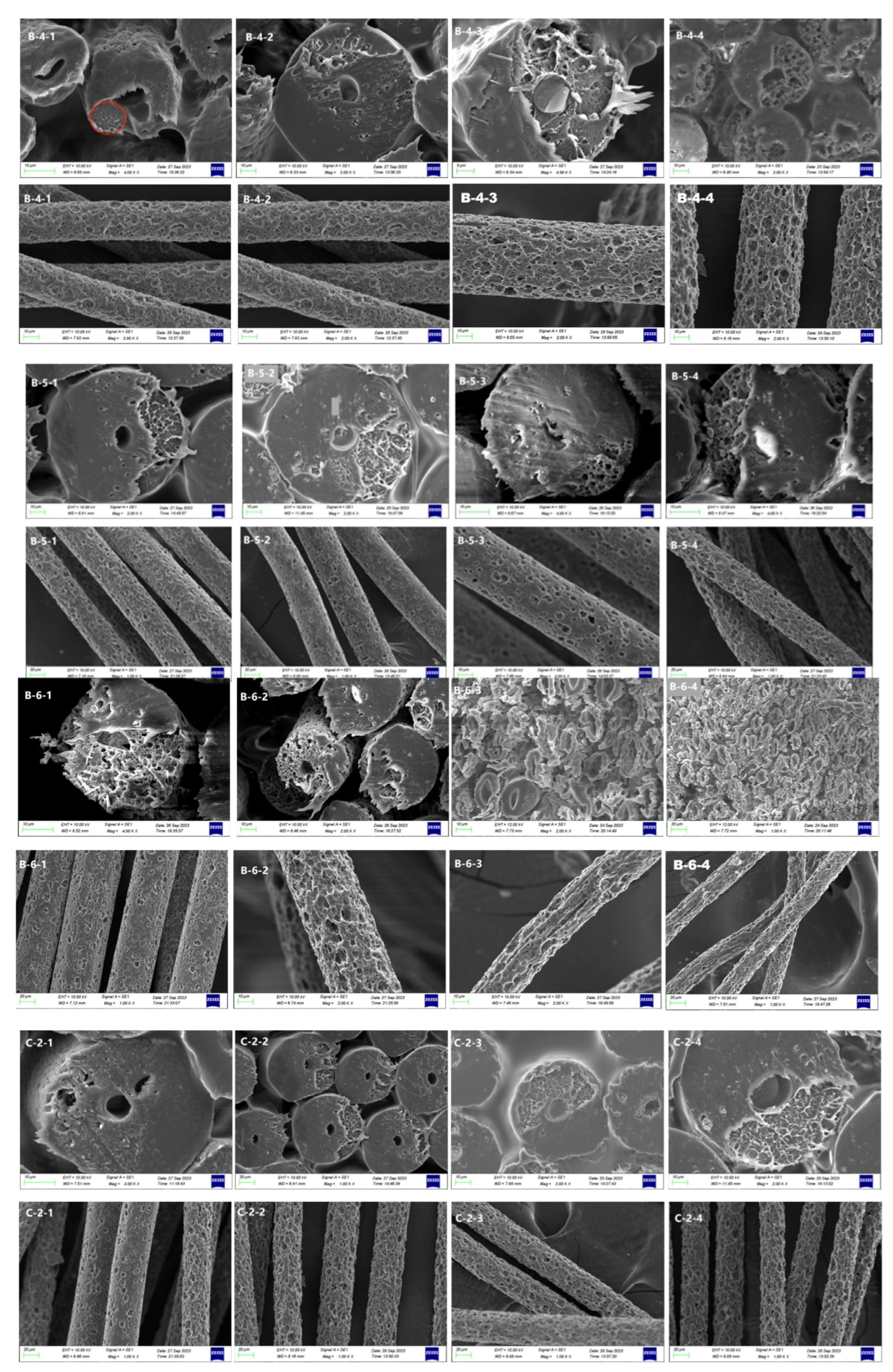
3.2. Morphological Structure
3.2.1. Scanning Electron Microscopy after Alkali Treatment
3.2.2. Scanning Electron Microscopy after Acid Treatment
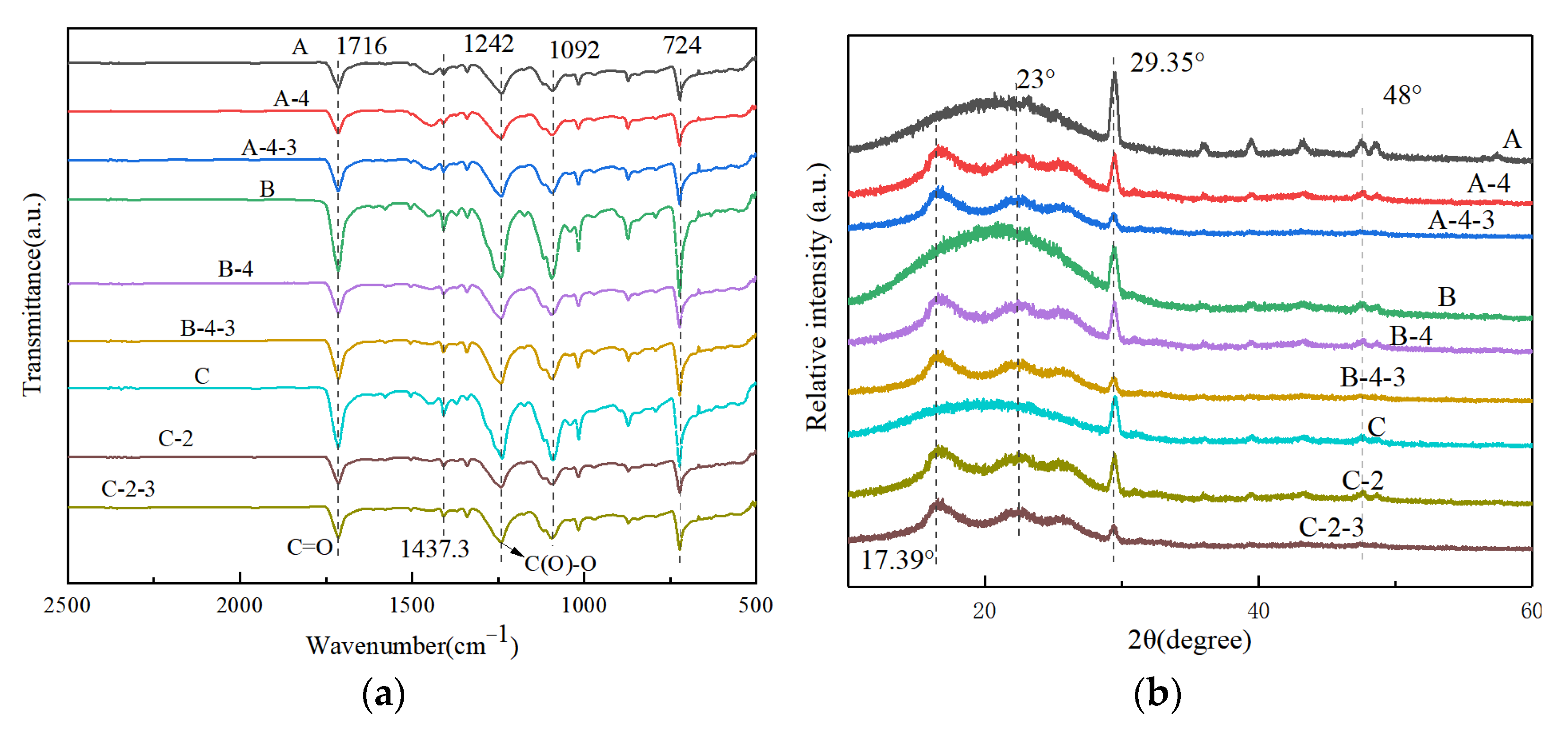
3.3. Chemical Structure and Crystalline Property
3.4. Thermal Analysis
3.5. Strength and Linear Density
3.5.1. Strength and Density after Alkali Treatment
3.5.2. Strength and Density of Monofilament after Acid Treatment
3.6. Porous Structure
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Costantini, M.; Colosi, C.; Święszkowski, W.; Barbetta, A. Co-axial wet-spinning in 3D bioprinting: State of the art and future perspective of microfluidic integration. Biofabrication 2018, 11, 012001. [Google Scholar] [CrossRef] [PubMed]
- Rihova, M.; Ince, A.E.; Cicmancova, V.; Hromadko, L.; Castkova, K.; Pavlinak, D.; Vojtova, L.; Macak, J.M. Water-born 3D nanofiber mats using cost-effective centrifugal spinning: Comparison with electrospinning process: A complex study. J. Appl. Polym. Sci. 2021, 138, 49975. [Google Scholar] [CrossRef]
- Aydin, L.; Karatoprak, A.P.; Kucuk, S. Biologic: H-Bot Kinematics Based Multi-Micro-Extrusion Bioprinter. J. Med. Devices 2023, 17, 020902. [Google Scholar] [CrossRef]
- Penconek, A.; Jackiewicz-Zagórska, A.; Przekop, R.; Moskal, A. Fibrous Structures Produced Using the Solution Blow-Spinning Technique for Advanced Air Filtration Process. Materials 2023, 16, 7118. [Google Scholar] [CrossRef] [PubMed]
- An, Y.X.; Li, P.L.; Wu, H.Y. Preparation of microporous poly(vinylidene fluoride)hollow fiber membranes via thermally induced phase separation. Membr. Sci. Technol. 2013, 33, 13–19. [Google Scholar]
- Jafari, D.; van Alphen, K.J.; Geurts, B.J.; Wits, W.W.; Gonzalez, L.C.; Vaneker, T.H.; Rahman, N.U.; Römer, G.W.; Gibson, I. Porous materials additively manufactured at low energy: Single-layer manufacturing and characterization. Mater. Des. 2020, 191, 108654. [Google Scholar] [CrossRef]
- Aksoy, Y.; Hasar, H. Fabrication of gas-permeable polyvinylidene fluoride (PVDF) hollow-fiber membrane by dry-jet wet spinning and its application in membrane biofilm reactors. J. Water Process Eng. 2021, 40, 101879. [Google Scholar] [CrossRef]
- Sethunga, G.; Karahan, H.E.; Wang, R.; Bae, T.-H. PDMS-coated porous PVDF hollow fiber membranes for efficient recovery of dissolved biomethane from anaerobic effluents. J. Membr. Sci. 2019, 584, 333–342. [Google Scholar] [CrossRef]
- Radebe, L.; Wesley-Smith, J.; Focke, W.W.; Ramjee, S. Formulating calcium carbonate masterbatches. J. Polym. Eng. 2023, 43, 80–88. [Google Scholar] [CrossRef]
- Chafidz, A. Polypropylene/CaCO3 Nanocomposites Fabricated Using Masterbatch: Effect of Nano-CaCO3 Loadings and Re-Processing on the Melting Properties. Mater. Sci. Forum 2022, 1067, 73–78. [Google Scholar] [CrossRef]
- Xu, G.Q.; Li, X.F.; Lv, X.L. Calcium carbonate on the thermal induced phase separation and preparation of poly (vinylidene fluoride porous membrane structure. Influ. Polym. Mater. Sci. Eng. 2007, 5, 234–237. [Google Scholar] [CrossRef]
- Neelima, T.; Manjusri, M.; Amar, K. Durable polylactic acid (PLA)-based sustainable engineered blends and biocomposites: Recent developments, challenges, and opportunities. ACS Eng. Au 2021, 1, 7–38. [Google Scholar]
- Bahlouli, S.; Makhlouf, A.; Haddaoui, N. Influence of nanosized CaCO3 content in tailoring the structure, the morphology and the thermal and mechanical properties of iPP/PA66/PP-g-MA alloy. Int. J. Polym. Anal. Charact. 2021, 26, 440–457. [Google Scholar] [CrossRef]
- Qi, C.X. Preparation and Catalytic Performance of Composite Photocatalytic Fiber Based on Graphite Phase Carbon Nitride/Calcium Carbonate; Zhejiang University of Technology: Hangzhou, China, 2022. [Google Scholar]
- Loganathan, T.M.; Sultan, M.T.H.; Ahsan, Q.; Jawaid, M.; Naveen, J.; Shah, A.U.M.; Hua, L.S. Characterization of alkali treated new cellulosic fibre from Cyrtostachys renda. J. Mater. Res. Technol. 2020, 9, 3537–3546. [Google Scholar] [CrossRef]
- Ni, H.; Li, Y.; Chen, Z. Study on preparation and properties of luminescent polyamide fiber doped with graphene. J. Phys. Conf. Ser. 2020, 1622, 012038. [Google Scholar] [CrossRef]
- Wang, C.; Su, J.; Liu, T. A sustainable strategy to transform cotton waste into renewable cellulose fiber self-reinforcing composite paper. J. Clean. Prod. 2023, 429, 139567. [Google Scholar] [CrossRef]
- Zhao, H.; Guo, T.; Lu, Z. Genome-wide association studies detects candidate genes for wool traits by re-sequencing in Chinese fine-wool sheep. BMC Genom. 2021, 22, 1–13. [Google Scholar] [CrossRef]
- Li, X.W.; Yang, Y.; Lu, J.; Dong, C.; Wang, Y.G.; Xu, X.Z. Growth and properties of large size lithium terbium fluoride Crystals. J. Synth. Cryst. 2023, 52, 1352–1356. [Google Scholar]
- Zhai, C.H.; Feng, Y.H.; Ye, Z.G. Study on Synthesis of Polyester Resin for Powder Coatings with Outstanding Alkali Resistance. Coat. Prot./Tuceng Yu Fanghu 2023, 44, 34–38. [Google Scholar]
- Duan, P.; Lei, T.; Han, Y. Effect of warm-mixing wax’s molecular weights on microstructure, rheological and mechanical properties of asphalt composites. Constr. Build. Mater. 2023, 408, 133620. [Google Scholar] [CrossRef]
- Joubert, R.-L.; Jokonya, O. A systematic literature review of factors affecting the adoption of technologies in food waste management. Procedia Comput. Sci. 2021, 181, 1034–1040. [Google Scholar] [CrossRef]
- Alsaid, A.A. Alkaline hydrolysis of polyester woven fabrics and its influence on thermal comfort properties. Egypt. J. Chem. 2022, 65, 259–274. [Google Scholar]
- Modjinou, T.; Lemechko, P.; Babinot, J.; Versace, D.-L.; Langlois, V.; Renard, E. Poly (3-hydroxyalkanoate) sulfonate: From nanoparticles toward water soluble polyesters. Eur. Polym. J. 2015, 68, 471–479. [Google Scholar] [CrossRef]
- Muszyński, M.; Nowicki, J.; Zygadło, M.; Dudek, G. Comparsion of Catalyst Effectiveness in Different Chemical Depolymerization Methods of Poly(ethylene terephthalate). Molecules 2023, 28, 6385. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H. Alkaline hydrolysis and physical property changes of polyester interleaved with PET/COPET composite radiofiber as stomach yarn. Korean J. Fiber Eng. 1998, 35, 721–729. [Google Scholar]
- Li, G.; Gu, L.; Sun, T. Kinetics and mechanism relating to the alkali hydrolysis of polyester blend fibres. J. Donghua Univ. 1993, 2, 34. [Google Scholar]
- BXue, B.; Qin, R.; Shao, M.; Li, S.; Niu, M. Improving the flame retardancy of PET fiber by constructing the carbon microspheres based melamine polyphosphate powder. J. Text. Inst. 2020, 111, 597–603. [Google Scholar]
- Ardoña, H.A.M.; Paredes, F.U.; Arellano, I.H.J.; Arco, S.D. Electrospun PET supported-ionic liquid-stabilized CdS catalyst for the photodegradation of Rhodamine B under visible light. Mater. Lett. 2013, 91, 96–99. [Google Scholar] [CrossRef]
- Shen, X.H.; Wu, X.J.; Du, W.P. Preparation and characterization of electro-spinning Taiji stone masterbatch /PET composite fiber Shanghai Textile Science and Technology. Shanghai Text. Sci. Technol. 2016, 44, 47–50. [Google Scholar]
- Vagenas, N.V.; Gatsouli, A.; Kontoyannis, C.G. Quantitative analysis of synthetic calcium carbonate polymorphs using FT-IR spectroscopy. Talanta 2003, 59, 831–836. [Google Scholar] [CrossRef]
- Qin, D.; Lu, W.; Wang, X.; Li, N.; Chen, X.; Zhu, Z.; Chen, W. Graphitic Carbon Nitride from Burial to Re-emergence on Polyethylene Terephthalate Nanofibers as an Easily Recycled Photocatalyst for Degrading Antibiotics under Solar Irradiation. ACS Appl. Mater. Interfaces 2016, 8, 25962–25970. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Tang, X.; Wang, Z.; Peng, X. Pore characterization of different types of coal from coal and gas outburst disaster sites using low temperature nitrogen adsorption approach. Int. J. Min. Sci. Technol. 2017, 27, 371–377. [Google Scholar] [CrossRef]
- Sheng, Y. Study on Synthesis of Functional Nano-Calcium Carbonate Composites; Jilin University: Changchun, China, 2006. [Google Scholar]
- Aliotta, L.; Sciara, L.M.; Cinelli, P.; Canesi, I.; Lazzeri, A. Improvement of the PLA Crystallinity and Heat Distortion Temperature Optimizing the Content of Nucleating Agents and the Injection Molding Cycle Time. Polymers 2022, 14, 977. [Google Scholar] [CrossRef] [PubMed]
- Nooraiepour, M.; Masoudi, M.; Shokri, N.; Hellevang, H. Probabilistic Nucleation and Crystal Growth in Porous Medium: New Insights from Calcium Carbonate Precipitation on Primary and Secondary Substrates. ACS Omega 2021, 6, 28072–28083. [Google Scholar] [CrossRef] [PubMed]
- Ptiček Siročić, A.; Fijačko, A.; Hrnjak-Murgić, Z. Chemical recycling of postconsumer poly (ethylene-terephthalate) bottles–depolymerization study. Chem. Biochem. Eng. Q. 2013, 27, 65–71. [Google Scholar]
- Barhoum, A.; Rahier, H.; Abou-Zaied, R.E.; Rehan, M.; Dufour, T.; Hill, G.; Dufresne, A. Effect of Cationic and Anionic Surfactants on the Application of Calcium Carbonate Nanoparticles in Paper Coating. Carbon 2014, 36, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Karunadasa, K.S.; Manoratne, C.H.; Pitawala, H.M.T.G.A.; Rajapakse, R.M.G. Thermal decomposition of calcium carbonate (calcite polymorph) as examined by in-situ high-temperature X-ray powder diffraction. J. Phys. Chem. Solids 2019, 134, 21–28. [Google Scholar] [CrossRef]
- Lei, L.I.; He, H.; Wei, Y. Study on Properties of Polypropylene/Polyoxymethylene /CaCO3 Masterbatch Composite. Synth. Mater. Aging Appl. 2015, 44, 29–33. [Google Scholar] [CrossRef]
- Wilson, M.P.W. Shrinkage and chain folding in drawn poly (ethylene terephthalate) fibres. Polymer 1974, 15, 277–282. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.M. Study on shrinking property of high- shrinkage polyester staple fiber by chemical modification. Synth. Technol. Appl. 2016, 31, 13–16. [Google Scholar]
- Paz-Ferreiro, J.; Da Luz, L.R.Q.P.; Lado, M.; Vázquez, E.V. Specific surface area and multifractal parameters of associated nitrogen adsorption and desorption isotherms in soils from Santa Catarina, Brazil. Vadose Zone J. 2013, 12, vzj2012-0203. [Google Scholar] [CrossRef]
- Gregg, S.J. Adsorption, Specific Surface and Porosity; Chemical Industry Press: Beijing, China, 1989. [Google Scholar]
- Thomas, W.J.; Crittenden, B. Adsorption Technology and Design; Butterworth-Heinemann: Oxford, UK, 1998. [Google Scholar]
- Mietek, J.; Solovyov, L.A. Improvement of the Kruk–Jaroniec–Sayari method for pore size analysis of ordered silicas with cylindrical mesopores. Langmuir 2006, 22, 6757–6760. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Tm (°C) | Tc (°C) | ΔHm (J/g) | Xc (%) |
|---|---|---|---|---|
| A | 255.94 | 239.20 | 55.39 | 39.56 |
| A-4 | 255.18 | 238.19 | 51.42 | 36.73 |
| A-4-3 | 255.08 | 238.87 | 40.70 | 29.07 |
| B | 253.29 | 240.01 | 64.19 | 41.85 |
| B-4 | 253.86 | 237.78 | 61.68 | 40.06 |
| B-4-3 | 253.91 | 238.87 | 38.34 | 27.39 |
| C | 252.85 | 239.09 | 73.67 | 42.62 |
| C-2 | 252.14 | 238.18 | 70.71 | 40.50 |
| C-2-3 | 252.41 | 239.84 | 21.66 | 15.47 |
| Yarn Name | Linear Density/dtex | Breaking Strength/cN/dtex | Elongation at Break/% |
|---|---|---|---|
| A-0 | 4.91 | 2.24 | 232 |
| A-1 | 20.66 | 0.42 | 209 |
| A-2 | 19.57 | 0.42 | 152 |
| A-3 | 16.87 | 0.38 | 117 |
| A-4 | 14.64 | 0.31 | 101 |
| A-5 | 12.32 | 0.30 | 98 |
| A-6 | 9.95 | 0.19 | 73 |
| B-0 | 5.05 | 1.84 | 198 |
| B-1 | 23.03 | 0.21 | 137 |
| B-2 | 21.63 | 0.20 | 109 |
| B-3 | 19.07 | 0.19 | 99 |
| B-4 | 16.94 | 0.18 | 91 |
| B-5 | 15.75 | 0.16 | 49 |
| B-6 | 6.95 | 0.15 | 34 |
| C-0 | 5.23 | 1.85 | 110 |
| C-1 | 24.56 | 0.14 | 89 |
| C-2 | 20.37 | 0.14 | 72 |
| C-3 | 9.88 | 0.11 | 20 |
| C-4 | 5.81 | 0.05 | 16 |
| C-5 | 0.72 | - | - |
| C-6 | - | - | - |
| Yarn Name | Linear Density/dtex | Breaking Strength/cN/dtex | Elongation at Break/% |
|---|---|---|---|
| A-1-1 | 19.37 | 0.40 | 201 |
| A-1-2 | 18.95 | 0.38 | 193 |
| A-1-3 | 17.97 | 0.36 | 181 |
| A-1-4 | 16.44 | 0.36 | 141 |
| A-2-1 | 18.69 | 0.36 | 155 |
| A-2-2 | 17.83 | 0.32 | 122 |
| A-2-3 | 16.81 | 0.30 | 98 |
| A-2-4 | 15.95 | 0.28 | 85 |
| A-3-1 | 16.21 | 0.36 | 105 |
| A-3-2 | 15.78 | 0.33 | 97 |
| A-3-3 | 14.69 | 0.32 | 83 |
| A-3-4 | 13.17 | 0.30 | 56 |
| A-4-1 | 13.87 | 0.30 | 93 |
| A-4-2 | 12.42 | 0.26 | 59 |
| A-4-3 | 11.76 | 0.23 | 47 |
| A-4-4 | 10.94 | 0.19 | 32 |
| A-5-1 | 11.94 | 0.30 | 92 |
| A-5-2 | 10.83 | 0.28 | 63 |
| A-5-3 | 10.32 | 0.28 | 35 |
| A-5-4 | 9.57 | 0.21 | 21 |
| A-6-1 | 9.13 | 0.12 | 68 |
| A-6-2 | 8.74 | 0.09 | 57 |
| A-6-3 | 8.02 | 0.07 | 31 |
| A-6-4 | 7.63 | 0.07 | 12 |
| B-1-1 | 22.76 | 0.18 | 126 |
| B-1-2 | 21.37 | 0.15 | 109 |
| B-1-3 | 20.45 | 0.14 | 94 |
| B-1-4 | 18.39 | 0.13 | 47 |
| B-2-1 | 20.01 | 0.19 | 89 |
| B-2-2 | 19.32 | 0.17 | 64 |
| B-2-3 | 18.54 | 0.15 | 41 |
| B-2-4 | 17.31 | 0.11 | 21 |
| B-3-1 | 18.74 | 0.17 | 84 |
| B-3-2 | 18.09 | 0.16 | 65 |
| B-3-3 | 17.31 | 0.12 | 37 |
| B-3-4 | 16.84 | 0.11 | 17 |
| B-4-1 | 12.87 | 0.24 | 83 |
| B-4-2 | 12.03 | 0.22 | 62 |
| B-4-3 | 11.21 | 0.18 | 51 |
| B-4-4 | 10.83 | 0.17 | 32 |
| B-5-1 | 9.37 | 0.22 | 45 |
| B-5-2 | 7.92 | 0.20 | 34 |
| B-5-3 | 7.01 | 0.15 | 27 |
| B-5-4 | 6.34 | 0.15 | 15 |
| B-6-1 | 5.96 | 0.33 | 30 |
| B-6-2 | 5.01 | 0.28 | 21 |
| B-6-3 | 4.82 | 0.22 | 20 |
| B-6-4 | 4.03 | 0.19 | 17 |
| C-1-1 | 22.38 | 0.14 | 76 |
| C-1-2 | 21.08 | 0.11 | 62 |
| C-1-3 | 20.05 | 0.10 | 43 |
| C-1-4 | 19.13 | 0.05 | 18 |
| C-2-1 | 18.76 | 0.10 | 61 |
| C-2-2 | 17.01 | 0.07 | 41 |
| C-2-3 | 16.54 | 0.06 | 24 |
| C-2-4 | 15.13 | 0.05 | 19 |
| C-3-1 | 8.25 | 0.11 | 20 |
| C-3-2 | 7.83 | 0.09 | 17 |
| C-3-3 | 6.92 | 0.08 | 15 |
| C-3-4 | 5.73 | 0.08 | 9 |
| C-4-1 | 5.02 | 0.06 | 14 |
| C-4-2 | 4.71 | 0.05 | 12 |
| C-4-3 | 2.02 | 0.05 | 8 |
| C-4-4 | 1.09 | 0.07 | 5 |
| C-5-1 | - | - | - |
| C-5-2 | - | - | - |
| C-5-3 | - | - | - |
| C-5-4 | - | - | - |
| C-6-1 | - | - | - |
| C-6-2 | - | - | - |
| C-6-3 | - | - | - |
| C-6-4 | - | - | - |
| Sample | BET Specific Surface Area (m2/g) | BET Pore Volume (cm3/g) | Mean Aperture (nm) |
|---|---|---|---|
| A | 0.44 | 0.004 | 6.48 |
| A-4 | 7.00 | 0.003 | 11.27 |
| A-4-3 | 5.52 | 0.002 | 10.87 |
| B | 0.88 | 0.005 | 6.53 |
| B-4 | 4.50 | 0.002 | 7.82 |
| B-4-3 | 1.52 | 0.001 | 2.13 |
| C | 0.96 | 0.004 | 7.09 |
| C-2 | 2.30 | 0.002 | 7.06 |
| C-2-3 | 0.41 | 0.001 | 1.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Song, R.; Pan, R.; Zhang, M.; Liu, L. Preparation and Characterization of Calcium Carbonate Masterbatch–Alkali Soluble Polyester/Polyester Porous Fiber via Melt Spinning. Materials 2024, 17, 160. https://doi.org/10.3390/ma17010160
Zhao Y, Song R, Pan R, Zhang M, Liu L. Preparation and Characterization of Calcium Carbonate Masterbatch–Alkali Soluble Polyester/Polyester Porous Fiber via Melt Spinning. Materials. 2024; 17(1):160. https://doi.org/10.3390/ma17010160
Chicago/Turabian StyleZhao, Yanjiao, Ruochen Song, Runan Pan, Meiling Zhang, and Lifang Liu. 2024. "Preparation and Characterization of Calcium Carbonate Masterbatch–Alkali Soluble Polyester/Polyester Porous Fiber via Melt Spinning" Materials 17, no. 1: 160. https://doi.org/10.3390/ma17010160
APA StyleZhao, Y., Song, R., Pan, R., Zhang, M., & Liu, L. (2024). Preparation and Characterization of Calcium Carbonate Masterbatch–Alkali Soluble Polyester/Polyester Porous Fiber via Melt Spinning. Materials, 17(1), 160. https://doi.org/10.3390/ma17010160