
Goldene: An Anisotropic Metallic Monolayer with Remarkable Stability and Rigidity and Low Lattice Thermal Conductivity


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Computational Methods
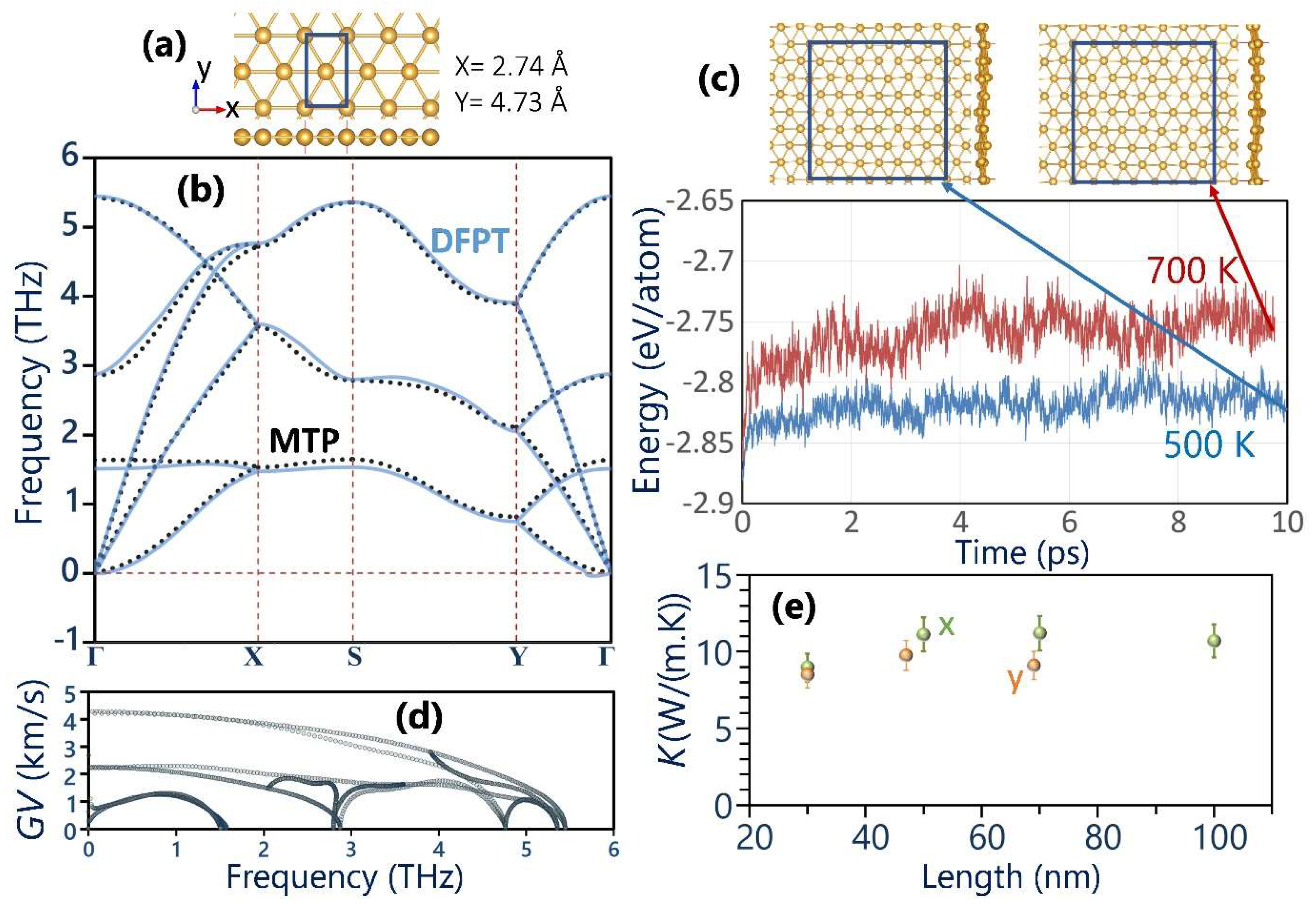
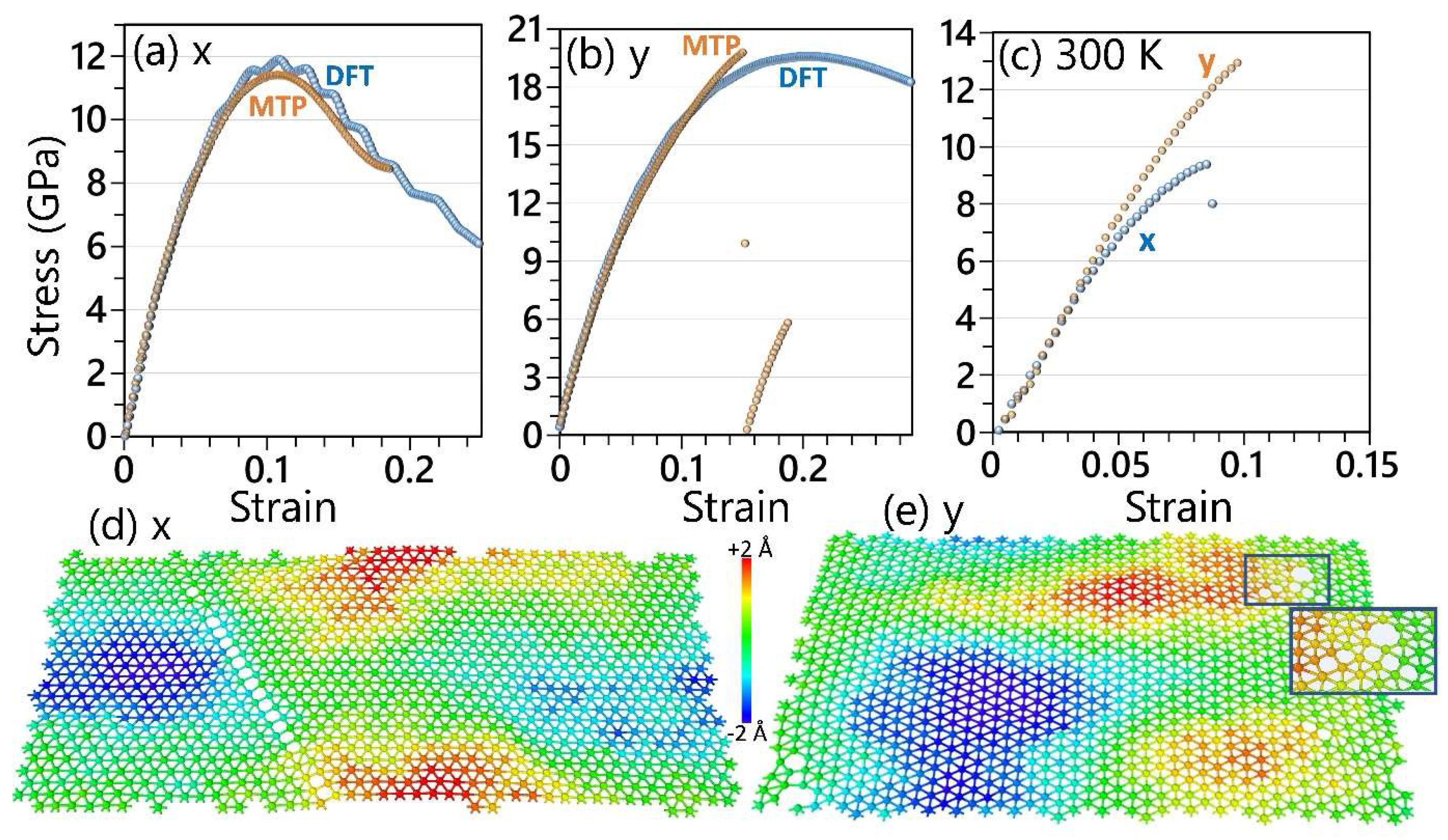
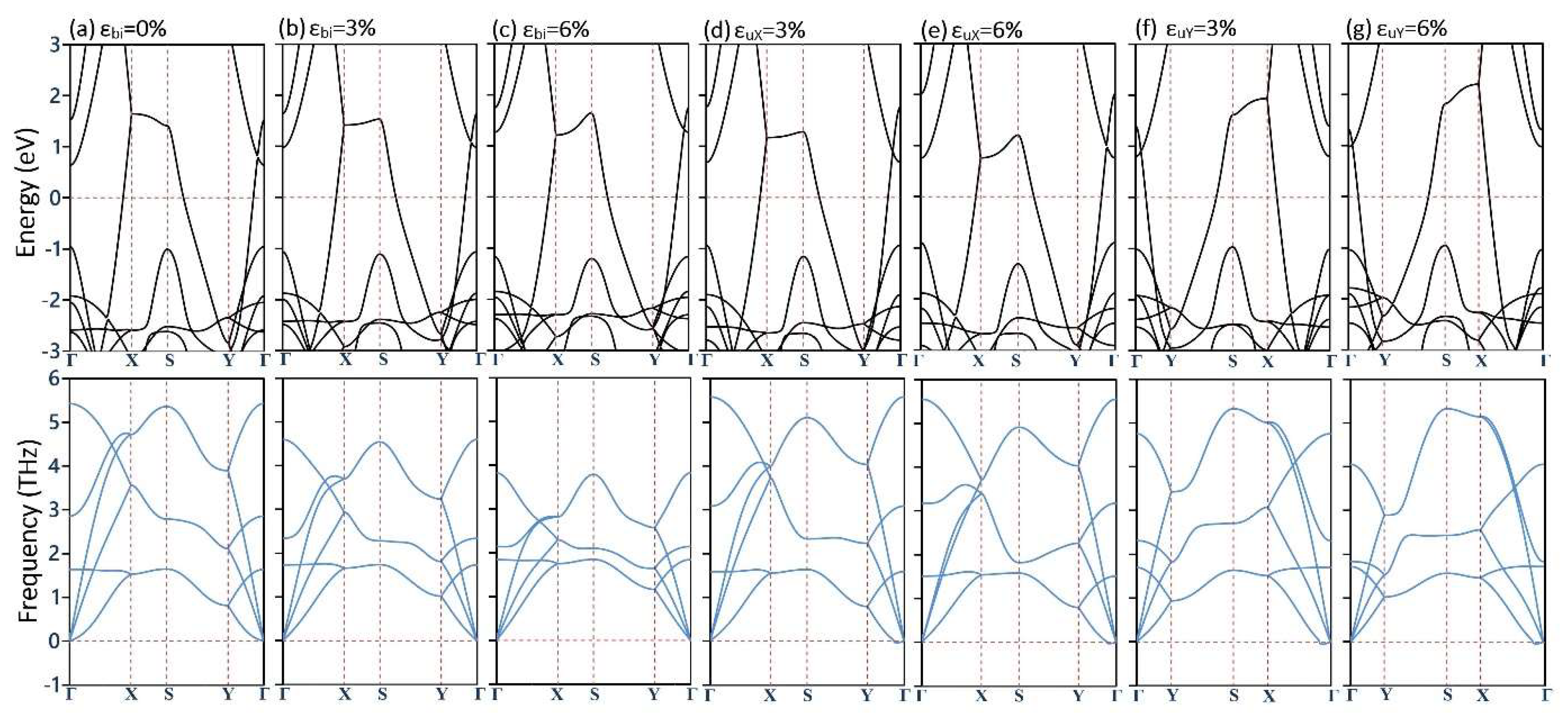
3. Results and Discussions
4. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Neto, A.H.C.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K.; Guinea, F. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef]
- Mannix, A.J.; Zhou, X.-F.; Kiraly, B.; Wood, J.D.; Alducin, D.; Myers, B.D.; Liu, X.; Fisher, B.L.; Santiago, U.; Guest, J.R.; et al. Synthesis of borophenes: Anisotropic, two-dimensional boron polymorphs. Science 2015, 350, 1513–1516. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Zhang, J.; Zhong, Q.; Li, W.; Li, S.; Li, H.; Cheng, P.; Meng, S.; Chen, L.; Wu, K. Experimental realization of two-dimensional boron sheets. Nat. Chem. 2016, 8, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Guo, S.; Chen, Z.; Wang, Y.; Gao, H.; Gómez-Herrero, J.; Ares, P.; Zamora, F.; Zhu, Z.; Zeng, H. Recent progress in 2D group-VA semiconductors: From theory to experiment. Chem. Soc. Rev. 2018, 47, 982–1021. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Neal, A.T.; Zhu, Z.; Luo, Z.; Xu, X.; Tománek, D.; Ye, P.D. Phosphorene: An Unexplored 2D Semiconductor with a High Hole Mobility. ACS Nano 2014, 8, 4033–4041. [Google Scholar] [CrossRef]
- Lin, H.; Qiu, W.; Liu, J.; Yu, L.; Gao, S.; Yao, H.; Chen, Y. Silicene: Wet-chemical exfoliation synthesis and biodegradable tumor nanomedicine. Adv. Mater. 2019, 31, 1903013. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Chen, W.; Xu, Y.; Gao, C.; Guan, D.; Liu, C. Epitaxial Growth of Two-Dimensional Stanene. Nat. Mater. 2015, 14, 1020–1025. [Google Scholar] [CrossRef]
- Miró, P.; Audiffred, M.; Heine, T. An atlas of two-dimensional materials. Chem. Soc. Rev. 2014, 43, 6537–6554. [Google Scholar] [CrossRef]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the Elastic Properties and Intrinsic Strength of Monolayer Graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Calizo, I.; Teweldebrhan, D.; Pokatilov, E.P.; Nika, D.L.; Balandin, A.A.; Bao, W.; Miao, F.; Lau, C.N. Extremely high thermal conductivity of graphene: Prospects for thermal management applications in nanoelectronic circuits. Appl. Phys. Lett. 2008, 92, 151911. [Google Scholar] [CrossRef]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior thermal conductivity of single-layer graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef]
- Berger, C.; Song, Z.; Li, T.; Li, X.; Ogbazghi, A.Y.; Feng, R.; Dai, Z.; Marchenkov, A.N.; Conrad, E.H.; First, P.N.; et al. Ultrathin Epitaxial Graphite: 2D Electron Gas Properties and a Route toward Graphene-based Nanoelectronics. J. Phys. Chem. B 2004, 108, 19912–19916. [Google Scholar] [CrossRef]
- Liu, M.; Yin, X.; Ulin-Avila, E.; Geng, B.; Zentgraf, T.; Ju, L.; Wang, F.; Zhang, X. A graphene-based broadband optical modulator. Nature 2011, 474, 64–67. [Google Scholar] [CrossRef]
- Withers, F.; Dubois, M.; Savchenko, A.K. Electron properties of fluorinated single-layer graphene transistors. Phys. Rev. B Condens. Matter Mater. Phys. 2010, 82, 073403. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, K. Recent progress on graphene-analogous 2D nanomaterials: Properties, modeling and applications. Prog. Mater. Sci. 2019, 100, 99–169. [Google Scholar] [CrossRef]
- Mortazavi, B.; Podryabinkin, E.V.; Novikov, I.S.; Rabczuk, T.; Zhuang, X.; Shapeev, A.V. Accelerating first-principles estimation of thermal conductivity by machine-learning interatomic potentials: A MTP/ShengBTE solution. Comput. Phys. Commun. 2021, 258, 107583. [Google Scholar] [CrossRef]
- Penev, E.S.; Bhowmick, S.; Sadrzadeh, A.; Yakobson, B.I. Polymorphism of two-dimensional boron. Nano Lett. 2012, 12, 2441–2445. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, Y.; Gao, G.; Yakobson, B.I. Two-Dimensional Boron Monolayers Mediated by Metal Substrates. Angew. Chem. 2015, 127, 13214–13218. [Google Scholar] [CrossRef]
- Zhou, X.F.; Dong, X.; Oganov, A.R.; Zhu, Q.; Tian, Y.; Wang, H.T. Semimetallic two-dimensional boron allotrope with massless Dirac fermions. Phys. Rev. Lett. 2014, 112, 085502. [Google Scholar] [CrossRef]
- Jiang, H.R.; Lu, Z.; Wu, M.C.; Ciucci, F.; Zhao, T.S. Borophene: A promising anode material offering high specific capacity and high rate capability for lithium-ion batteries. Nano Energy 2016, 23, 97–104. [Google Scholar] [CrossRef]
- Mortazavi, B.; Rahaman, O.; Ahzi, S.; Rabczuk, T. Flat borophene films as anode materials for Mg, Na or Li-ion batteries with ultra high capacities: A first-principles study. Appl. Mater. Today 2017, 8, 60–67. [Google Scholar] [CrossRef]
- Batmunkh, M.; Bat-erdene, M.; Shapter, J.G. Phosphorene and phosphorene-based materials—Prospects for future applications. Adv. Mater. 2016, 28, 8586–8617. [Google Scholar] [CrossRef]
- Kou, L.; Chen, C.; Smith, S.C. Phosphorene: Fabrication, Properties, and Applications. J. Phys. Chem. Lett. 2015, 6, 2794–2805. [Google Scholar] [CrossRef]
- Kashiwaya, S.; Shi, Y.; Lu, J.; Sangiovanni, D.G.; Greczynski, G.; Magnuson, M.; Andersson, M.; Rosen, J.; Hultman, L. Synthesis of goldene comprising single-atom layer gold. Nat. Synth. 2024, 1–8. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kresse, G.; Daniel, J. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Chadi, D.J.; Cohen, M.L. Special points in the brillouin zone. Phys. Rev. B 1973, 8, 5747–5753. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Shapeev, A.V. Moment tensor potentials: A class of systematically improvable interatomic potentials. Multiscale Model. Simul. 2016, 14, 1153–1173. [Google Scholar] [CrossRef]
- Mortazavi, B.; Silani, M.; Podryabinkin, E.V.; Rabczuk, T.; Zhuang, X.; Shapeev, A.V. First-Principles Multiscale Modeling of Mechanical Properties in Graphene/Borophene Heterostructures Empowered by Machine-Learning Interatomic Potentials. Adv. Mater. 2021, 33, 2102807. [Google Scholar] [CrossRef]
- Mortazavi, B.; Shojaei, F.; Shapeev, A.V.; Zhuang, X. A combined first-principles and machine-learning investigation on the stability, electronic, optical, and mechanical properties of novel C6N7-based nanoporous carbon nitrides. Carbon N. Y. 2022, 194, 230–239. [Google Scholar] [CrossRef]
- Mortazavi, B.; Zhuang, X.; Rabczuk, T.; Shapeev, A.V. Atomistic modeling of the mechanical properties: The rise of machine learning interatomic potentials. Mater. Horiz. 2023, 10, 1956–1968. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, B.; Novikov, I.S.; Podryabinkin, E.V.; Roche, S.; Rabczuk, T.; Shapeev, A.V.; Zhuang, X. Exploring phononic properties of two-dimensional materials using machine learning interatomic potentials. Appl. Mater. Today 2020, 20, 100685. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO—The Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2009, 18, 015012. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Mortazavi, B.; Podryabinkin, E.V.; Roche, S.; Rabczuk, T.; Zhuang, X.; Shapeev, A.V. Machine-learning interatomic potentials enable first-principles multiscale modeling of lattice thermal conductivity in graphene/borophene heterostructures. Mater. Horiz. 2020, 7, 2359–2367. [Google Scholar] [CrossRef]
- Li, J.; Guo, M.; Si, J.; Shi, L.; Shi, X.; Ma, J.-J.; Zhang, Q.; Singh, D.J.; Liu, P.-F.; Wang, B.-T. Coexistence of superconductivity and topological aspects in beryllenes. Mater. Today Phys. 2023, 38, 101257. [Google Scholar] [CrossRef]
- Mortazavi, B. Structural, electronic, thermal and mechanical properties of C60-based fullerene two-dimensional networks explored by first-principles and machine learning. Carbon N. Y. 2023, 213, 118293. [Google Scholar] [CrossRef]
- Wang, B.; Ying, P.; Zhang, J. The thermoelastic properties of monolayer covalent organic frameworks studied by machine-learning molecular dynamics. Nanoscale 2024, 16, 237–248. [Google Scholar] [CrossRef]
- Dong, H.; Shi, Y.; Ying, P.; Xu, K.; Liang, T.; Wang, Y.; Zeng, Z.; Wu, X.; Zhou, W.; Xiong, S.; et al. Molecular dynamics simulations of heat transport using machine-learned potentials: A mini-review and tutorial on GPUMD with neuroevolution potentials. J. Appl. Phys. 2024, 135, 161101. [Google Scholar] [CrossRef]
- Brorsson, J.; Hashemi, A.; Fan, Z.; Fransson, E.; Eriksson, F.; Ala-Nissila, T.; Krasheninnikov, A.V.; Komsa, H.-P.; Erhart, P. Efficient Calculation of the Lattice Thermal Conductivity by Atomistic Simulations with Ab Initio Accuracy. Adv. Theory Simul. 2022, 5, 2100217. [Google Scholar] [CrossRef]
- Cui, C.; Zhang, Y.; Ouyang, T.; Chen, M.; Tang, C.; Chen, Q.; He, C.; Li, J.; Zhong, J. On-the-fly machine learning potential accelerated accurate prediction of lattice thermal conductivity of metastable silicon crystals. Phys. Rev. Mater. 2023, 7, 33803. [Google Scholar] [CrossRef]
- Han, J.; Zeng, Q.; Chen, K.; Yu, X.; Dai, J. Lattice Thermal Conductivity of Monolayer InSe Calculated by Machine Learning Potential. Nanomaterials 2023, 13, 1576. [Google Scholar] [CrossRef]
- Qiu, R.; Zeng, Q.; Wang, H.; Kang, D.; Yu, X.; Dai, J. Anomalous Thermal Transport across the Superionic Transition in Ice. Chin. Phys. Lett. 2023, 40, 116301. [Google Scholar] [CrossRef]
- Izadifar, M.; Valencia, N.C.; Xiao, P.; Ukrainczyk, N.; Koenders, E. 3D Off-Lattice Coarse-Grained Monte Carlo Simulations for Nucleation of Alkaline Aluminosilicate Gels. Materials 2023, 16, 1863. [Google Scholar] [CrossRef] [PubMed]
- Izadifar, M.; Ukrainczyk, N.; Koenders, E. Silicate Dissolution Mechanism from Metakaolinite Using Density Functional Theory. Nanomaterials 2023, 13, 1196. [Google Scholar] [CrossRef]
- Santos, E.A.J.; Lima, K.A.L.; Junior, L.A.R. Proposing TODD-graphene as a novel porous 2D carbon allotrope designed for superior lithium-ion battery efficiency. Sci. Rep. 2024, 14, 6202. [Google Scholar] [CrossRef] [PubMed]
- Lashkarara, S.; Fazlali, A.; Ghaseminezhad, K.; Fleck, C.; Salavati, M. Mechanism of plasma electrolytic oxidation in Mg3ZnCa implants: A study of double-layer formation and properties through nanoindentation. Sci. Rep. 2024, 14, 7380. [Google Scholar] [CrossRef] [PubMed]
- Santos, E.A.J.; Lima, K.A.L.; Mendonça, F.L.L.; da Silva, D.A.; Giozza, W.F.; Junior, L.A.R. PHOTH-graphene: A new 2D carbon allotrope with low barriers for Li-ion mobility. Sci. Rep. 2024, 14, 9526. [Google Scholar] [CrossRef] [PubMed]
- Salavati, M. Mechanical Properties of α-Chitin and Chitosan Biocomposite: A Molecular Dynamic Study. J. Compos. Sci. 2023, 7, 464. [Google Scholar] [CrossRef]
- Kanegae, G.B.; Junior, M.L.P.; Galvão, D.S.; Junior, L.A.R.; Fonseca, A.F. Enhanced Elastocaloric Effects in γ-Graphyne. ACS Appl. Mater. Interfaces 2024. [Google Scholar] [CrossRef] [PubMed]
- Mehr, F.R.; Kamrani, S.; Fleck, C.; Salavati, M. Global and Local Deformation Analysis of Mg-SiC Nanocomposites: Digital Image Correlation (DIC) and Representative Volume Element (RVE) Techniques. J. Compos. Sci. 2024, 8, 1. [Google Scholar] [CrossRef]
- Kuc, A.; Zibouche, N.; Heine, T. Influence of quantum confinement on the electronic structure of the transition metal sulfide TS2. Phys. Rev. B 2011, 83, 245213. [Google Scholar] [CrossRef]
- Arnold, F.M.; Ghasemifard, A.; Kuc, A.; Kunstmann, J.; Heine, T. Relaxation effects in twisted bilayer molybdenum disulfide: Structure, stability, and electronic properties. 2D Mater. 2023, 10, 45010. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mortazavi, B. Goldene: An Anisotropic Metallic Monolayer with Remarkable Stability and Rigidity and Low Lattice Thermal Conductivity. Materials 2024, 17, 2653. https://doi.org/10.3390/ma17112653
Mortazavi B. Goldene: An Anisotropic Metallic Monolayer with Remarkable Stability and Rigidity and Low Lattice Thermal Conductivity. Materials. 2024; 17(11):2653. https://doi.org/10.3390/ma17112653
Chicago/Turabian StyleMortazavi, Bohayra. 2024. "Goldene: An Anisotropic Metallic Monolayer with Remarkable Stability and Rigidity and Low Lattice Thermal Conductivity" Materials 17, no. 11: 2653. https://doi.org/10.3390/ma17112653