

First-Principles Calculate the Stability, Mechanical Properties and Electronic Structure of Carbide MC, M2C and M6C in M50NiL Steel
Abstract
1. Introduction
2. Computational Details
3. Results and Discussion
3.1. Crystal Parameters and Stability
3.2. Mechanical Properties
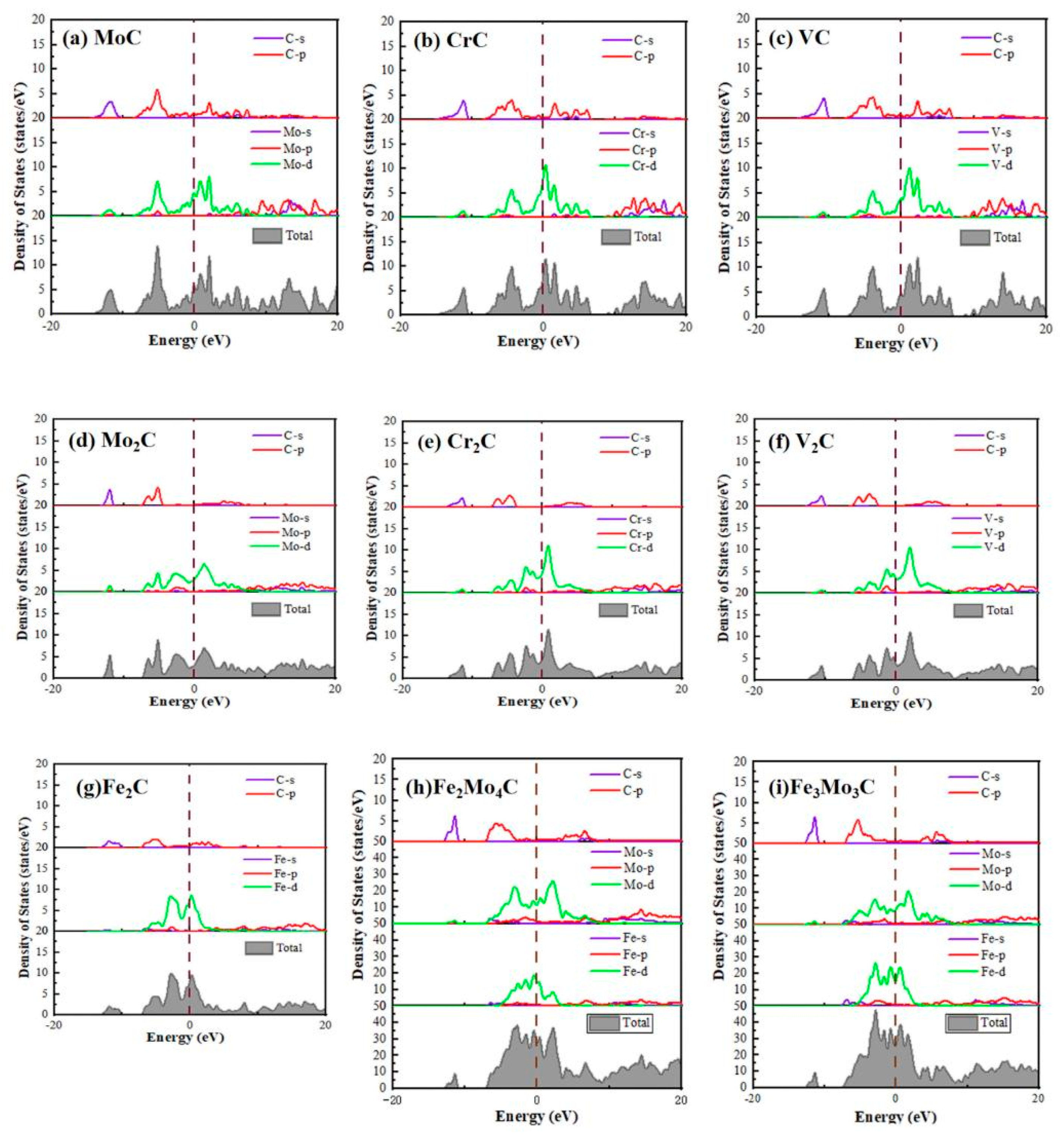
3.3. Electronic Structure Analysis
4. Conclusions
- (1)
- The binding energies of all phases are less than 0, indicating that these phases are stable. In the same phase, the stability of the second phase is related to the electron loss ability of its metal elements. The stronger the electron loss ability of its metal elements, the more stable the formed phase. The stability order of MC carbides is VC > MoC > CrC, while the stability order of M2C carbides is Mo2C > V2C > Cr2C > Fe2C. As for M6C carbides, the stability order is Fe2Mo4C > Fe3Mo3C.
- (2)
- Each phase’s hardness, ductility and anisotropy were analyzed by calculating the elastic modulus and anisotropy index . The hardness was measured by the Young’s modulus and the shear modulus. The greater the Young’s modulus and the shear modulus, the greater the hardness. For MC carbides, the hardness order is VC > CrC > MoC. As for M2C carbides, the hardness order is Mo2C > Cr2C > V2C > Fe2C. As for M6C carbides, the hardness order is Fe3Mo3C > Fe2Mo4C. The toughness and ductility order of all carbides is Fe2C > MoC > Fe2Mo4C > V2C > Fe3Mo3C > CrC > VC > Mo2C > Cr2C, and the anisotropy order of all carbides is MoC > V2C > VC > Cr2C > CrC > Mo2C > Fe2Mo4C > Fe3Mo3C.
- (3)
- The electronic properties of all carbides are analyzed. The results of the density of states and the charge density difference show that ionic bonds and covalent bonds will form in most carbides. The main reason is that the metal atom loses electrons, and the C atom obtains electrons. The stability of all carbides also conforms to the calculation results of binding energy.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bhadeshia, H.K.D.H. Steels for bearings. Prog. Mater. Sci. 2012, 57, 268–435. [Google Scholar]
- Wang, F.; Sun, Q.; Ren, H.; Cao, N.; Song, X.; Deng, S.; Qian, D.; Wu, M. A novel quenching-electroshocking-tempering process for toughness improvement by microstructure refining and austenite stability tailoring in aviation bearing steel. Mater. Sci. Eng. A Struct. 2022, 854, 143817. [Google Scholar] [CrossRef]
- Hetzner, D.W.; Van Geertruyden, W. Crystallography and metallography of carbides in high alloy steels. Mater. Charact. 2008, 59, 825–841. [Google Scholar] [CrossRef]
- Sun, L.; Li, M. High temperature behavior of isothermally compressed M50 steel. J. Iron Steel Res. Int. 2015, 22, 969–976. [Google Scholar] [CrossRef]
- Rosado, L.; Forster, N.H.; Thompson, K.L.; Cooke, J.W. Rolling contact fatigue life and spall propagation of AISI M50, M50NiL, and AISI 52100, Part I: Experimental results. Tribol. Trans. 2009, 53, 29–41. [Google Scholar] [CrossRef]
- Ozbek, I. Mechanical properties and kinetics of borided AISI M50 bearing steel. Arab. J. Sci. Eng. 2014, 39, 5185–5192. [Google Scholar] [CrossRef]
- Arakere, N.K.; Branch, N.; Levesque, G.; Svendsen, V.; Forster, N.H. Rolling contact fatigue life and spall propagation of AISI M50, M50NiL, and AISI 52100, Part II: Stress modeling. Tribol. Trans. 2009, 53, 42–51. [Google Scholar] [CrossRef]
- Ding, Z.; Guo, J.; Niu, J.; Zhou, L.; Zhang, X.; Ma, X. Fabrication and mechanical properties of micro/nano-crystalline layers in M50NiL carburized steel. Mater. Des. 2024, 241, 112888. [Google Scholar] [CrossRef]
- Li, G.; Liang, Y.; Sun, H.; Cao, Y.; Zhu, Z. Effect of pre-existing carbides prepared by different heat treatments on the nitriding behaviour during a carburizing and nitriding duplex treatment of an M50NiL steel. Surf. Coat. Technol. 2020, 395, 125930. [Google Scholar] [CrossRef]
- Yu, X.; Gao, Y.; Wang, S.; Wang, H.; Xia, Y.; Yang, S.; Su, Y. Effect of deep tempering on microstructure and me-chanical properties of G13Cr4Mo4Ni4V steel. Mater. Express 2021, 11, 1200–1206. [Google Scholar] [CrossRef]
- Du, N.; Liu, H.; Cao, Y.; Fu, P.; Sun, C.; Liu, H.; Li, D. Formation mechanism of MC and M2C primary carbides in as-cast M50 bearing steel. Mater. Charact. 2021, 174, 111011. [Google Scholar] [CrossRef]
- Du, N.; Liu, H.; Cao, Y.; Fu, P.; Sun, C.; Liu, H.; Li, D. In situ investigation of the fracture of primary carbides and its mechanism in M50 steel. Mater. Charact. 2022, 186, 111822. [Google Scholar] [CrossRef]
- YB/T 4106-2000; Carburizing Bearing Steel for High Temperature for Aircraft Engine. State Bureau of Metallurgical Industry: Beijing, China, 2000.
- Vogt, J.B. Fatigue properties of high nitrogen steels. J. Mater. Process. Technol. 2001, 117, 364–369. [Google Scholar] [CrossRef]
- Simmons, J.W. Overview: High-nitrogen alloying of stainless steels. Mater. Sci. Eng. A Struct. 1996, 207, 159–169. [Google Scholar] [CrossRef]
- Su, J.; Qian, D.; Wang, F. Effect of prior cold ring rolling on carbide dissolution during the austenitizing process of an M50 bearing steel. Mater. Express 2020, 10, 1010–1019. [Google Scholar] [CrossRef]
- Guo, J.; Liu, S.; Zhou, Y.; Wang, J.; Xing, X.; Ren, X.; Yang, Q. Stability of eutectic carbide in Fe-Cr-Mo-W-V-C alloy. Mater. Lett. 2016, 171, 216–219. [Google Scholar] [CrossRef]
- Jang, J.H.; Lee, C.H.; Heo, Y.U.; Suh, D.W. Stability of (Ti, M)C (M=Nb, V, Mo and W) carbide in steels using first-principles calculations. Acta Mater. 2012, 60, 208–217. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, M.; Ruan, L.; Zou, Z. A first-principles study on elastic properties and stability of TixV1-xC multiple carbide. Trans. Nonferrous Metal. Soc. 2011, 21, 1373–1377. [Google Scholar] [CrossRef]
- Suetin, D.V.; Medvedeva, N.I. Structural, electronic and magnetic properties of η-carbides M3W3C (M=Ti, V, Cr, Mn, Fe, Co, Ni). J. Alloys Compd. 2016, 681, 508–515. [Google Scholar] [CrossRef]
- Lv, Z.Q.; Wang, B.; Sun, S.H.; Fu, W.T. Effect of atomic sites on electronic and mechanical properties of (Fe,Mo)6C carbides. J. Alloys Compd. 2015, 649, 1089–1093. [Google Scholar] [CrossRef]
- Lian, J.; Zheng, L.; Wang, F.; Zhang, H. Evolution of carbides on surface of carburized M50NiL bearing steel. J. Iron Steel Res. Int. 2018, 25, 1198–1211. [Google Scholar] [CrossRef]
- Baerends, E.J. Perspective on “Self-consistent equations including exchange and correlation effects”. Theor. Chem. Acc. 2000, 103, 265–269. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Cao, T.; Cuffari, D.; Bongiorno, A. First-principles calculation of third-order elastic constants via numerical differentiation of the second Piola-Kirchhoff stress tensor. Phys. Rev. Lett. 2018, 121, 216001. [Google Scholar] [CrossRef]
- Ahmed, R.; Mahamudujjaman, M.; Afzal, M.A.; Islam, M.S.; Islam, R.S.; Naqib, S.H. DFT based comparative analysis of the physical properties of some binary transition metal carbides XC(X=Nb, Ta, Ti). J. Mater. Res. Technol. 2023, 24, 4808–4832. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, Y.; Feng, J.; Zhou, R. Elasticity, electronic properties and hardness of MoC investigated by first principles calculations. Physica B 2013, 419, 45–50. [Google Scholar] [CrossRef]
- Li, Y.; Gao, Y.; Xiao, B.; Min, T.; Yang, Y.; Ma, S.; Yi, D. The electronic, mechanical properties and theoretical hardness of chromium carbides by first-principles calculations. J. Alloys Compd. 2011, 509, 5242–5249. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, Y.; Zhou, R.; Feng, J. First principles study the stability and mechanical properties of MC(M=Ti, V, Zr, Nb, Hf and Ta) compounds. J. Alloys Compd. 2014, 582, 500–504. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, J.; Lai, Z.; Zhao, R.; He, D. A first-principles study on structural and electronic properties of Mo2C. Scripta Mater. 2009, 60, 949–952. [Google Scholar] [CrossRef]
- Wang, L.; Wen, J.; Jiang, Y.; Ou, Q.; Yu, L.; Xiong, B.S.; Yang, B.; Zhang, C.; Tong, Y. Electrical conduction characteristic of a 2D MXene device with Cu/Cr2C/TiN structure based on density functional theory. Materials 2020, 13, 3671. [Google Scholar] [CrossRef]
- Yu, R.; Chong, X.; Jiang, Y.; Zhou, R.; Yuan, W.; Feng, J. The stability, electronic structure, elastic and metallic properties of manganese nitrides. RSC Adv. 2015, 5, 1620–1627. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J.; Liang, W.; Gao, J.; Qi, Y.; Shang, C. Precipitation behaviors of carbides in high speed steel during ESR and heat treatment. Metals 2021, 11, 1781. [Google Scholar] [CrossRef]
- Zakeri, M.; Rahimipour, M.R.; Khanmohammadian, A. Mechanically activated synthesis of nanocrystalline ternary carbide Fe3Mo3C. Mater. Sci. Eng. A Struct. 2008, 492, 311–316. [Google Scholar] [CrossRef]
- Decaudin, B.; Djega-Mariadassou, C.; Cizeron, G. Structural study of M50 steel carbides. J. Alloys Compd. 1995, 226, 208–212. [Google Scholar] [CrossRef]
- Eberhart, M.E. The metallic bond: Elastic properties. Acta Mater. 1996, 44, 2495–2504. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Hu, W.C.; Liu, Y.; Li, D.J.; Zeng, X.Q.; Xu, C.S. First-principles study of structural and electronic properties of C14-type Laves phase Al2Zr and Al2Hf. Comp. Mater. Sci. 2014, 83, 27–34. [Google Scholar]
- Liu, X.; Yang, J.; Zhang, F.; Fu, X.; Li, H.; Yang, C. Experimental and DFT study on cerium inclusions in clean steels. J. Rare Earths 2021, 39, 477–486. [Google Scholar] [CrossRef]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal elastic anisotropy index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef]
- Ali, K.; Arya, A. Cohesive, elastic and anisotropic properties of s-, p- and d-block fission metals substituted Fe-Zr intermetallics. Prog. Nucl. Energy 2024, 168, 105025. [Google Scholar] [CrossRef]
- Yang, C.; Duan, Y.; Yu, J.; Peng, M.; Zheng, S.; Li, M. Elastic anisotropy and thermal properties of M-B-N (M=Al, Ga) systems using first-principles calculations. Vacuum 2023, 207, 111626. [Google Scholar] [CrossRef]
- Chen, K.; Zhao, L.R.; Rodgers, J.; Tse, J.S. Alloying effects on elastic properties of TiN-based nitrides. J. Phys. D Appl. Phys. 2003, 36, 2725–2729. [Google Scholar] [CrossRef]
- Ongwen, N.O.; Ogam, E.; Fellah, Z.E.A.; Otunga, H.O.; Oduor, A.O.; Mageto, M. Accurate Ab-initio calculation of elastic constants of anisotropic binary alloys: A case of Fe-Al. Solid State Commun. 2022, 353, 114879. [Google Scholar] [CrossRef]
- Chen, X.Q.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [Google Scholar] [CrossRef]
- Yan, R.; Chen, H.; Chen, Z.; Zhao, H.; Pan, F.; Wang, Y.; Qiao, J.; Liu, Z.; Huang, X.; Ma, X. Electronic structures, elastic and anisotropic properties of TiAl3 intermetallics doped with Nb. Physica B 2024, 688, 416158. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Mass Percentage/% | YB/T4106-2000 [13] |
|---|---|---|
| C | 0.147 | 0.13~0.15 |
| Cr | 4.14 | 4.00~4.25 |
| Mo | 4.28 | 4.00~4.50 |
| Ni | 3.37 | 3.20~3.60 |
| V | 1.17 | 1.13~1.33 |
| Nb | 0.02 | - |
| P | 0.003 | ≤0.008 |
| S | 0.002 | ≤0.003 |
| Carbides | Space Group | Lattice System | a (Å) | b (Å) | c (Å) | Volume (Å3) | |
|---|---|---|---|---|---|---|---|
| MC | MoC | cubic | 4.383 | - | - | 84.206 | |
| CrC | cubic | 4.108 | - | - | 69.330 | ||
| VC | cubic | 4.162 | - | - | 72.095 | ||
| M2C | Mo2C | hexagonal | 3.073 | 3.073 | 4.653 | 38.052 | |
| Cr2C | hexagonal | 2.833 | 2.833 | 4.259 | 29.603 | ||
| V2C | hexagonal | 2.893 | 2.893 | 4.532 | 32.849 | ||
| Fe2C | hexagonal | 3.699 | 3.699 | 2.642 | 31.307 | ||
| M6C | Fe2Mo4C | cubic | 8.016 | 8.016 | 8.016 | 364.214 | |
| Fe3Mo3C | cubic | 7.824 | 7.824 | 7.824 | 338.668 | ||
| Carbides | Enthalpy(eV) | Formation Enthalpy (eV/atom) | Binding Energy (eV/atom) | Reference | |
|---|---|---|---|---|---|
| MC | MoC | −8371.20 | −73.73 | −9.22 | −10.06 [27] |
| CrC | −10,495.99 | −69.46 | −8.68 | −9.56 [28] | |
| VC | −8534.52 | −71.97 | −9.00 | −8.1 [29] | |
| M2C | Mo2C | −8062.40 | −57.37 | −9.56 | - |
| Cr2C | −10,187.09 | −53.00 | −8.83 | - | |
| V2C | −8223.31 | −53.19 | −8.86 | - | |
| Fe2C | −3772.87 | −45.46 | −7.58 | - | |
| M6C | Fe2Mo4C | −38,566.18 | −261.65 | −9.34 | - |
| Fe3Mo3C | −34,285.21 | −258.38 | −9.23 | - | |
| Carbides | Fe | Cr | Mo | V |
|---|---|---|---|---|
| M6C | 35 | 7 | 56 | 2 |
| M2C | 3 | 13 | 69 | 15 |
| MC | 1 | 6 | 40 | 53 |
| Carbides | B (GPa) | G (GPa) | E (GPa) | B/G | |||
|---|---|---|---|---|---|---|---|
| MC | MoC | 355.97 | 85.86 | 238.42 | 0.39 | 4.15 | 2.87 |
| Ref. | 328.8 [27] | ||||||
| CrC | 322.24 | 170.32 | 434.41 | 0.28 | 1.89 | 0.26 | |
| Ref. | 317.7 [28] | 156.3 [28] | 402.8 [28] | 0.29 [28] | 2.03 [28] | ||
| VC | 305.19 | 178.08 | 447.24 | 0.26 | 1.71 | 0.34 | |
| M2C | Mo2C | 269.58 | 151.10 | 381.94 | 0.26 | 1.78 | 0.20 |
| Cr2C | 248.75 | 146.58 | 367.54 | 0.25 | 1.70 | 0.29 | |
| V2C | 217.68 | 102.96 | 266.80 | 0.30 | 2.11 | 2.12 | |
| Fe2C | 198.44 | 25.22 | 72.59 | 0.44 | 7.87 | - | |
| M6C | Fe2Mo4C | 282.98 | 125.58 | 328.20 | 0.31 | 2.25 | 0.001 |
| Fe3Mo3C | 295.12 | 144.92 | 373.61 | 0.29 | 2.04 | 0.000 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yong, X.; Liu, X.; Yang, M.; Zhou, X. First-Principles Calculate the Stability, Mechanical Properties and Electronic Structure of Carbide MC, M2C and M6C in M50NiL Steel. Materials 2024, 17, 3498. https://doi.org/10.3390/ma17143498
Yong X, Liu X, Yang M, Zhou X. First-Principles Calculate the Stability, Mechanical Properties and Electronic Structure of Carbide MC, M2C and M6C in M50NiL Steel. Materials. 2024; 17(14):3498. https://doi.org/10.3390/ma17143498
Chicago/Turabian StyleYong, Xi, Xiating Liu, Maosheng Yang, and Xiaolong Zhou. 2024. "First-Principles Calculate the Stability, Mechanical Properties and Electronic Structure of Carbide MC, M2C and M6C in M50NiL Steel" Materials 17, no. 14: 3498. https://doi.org/10.3390/ma17143498
APA StyleYong, X., Liu, X., Yang, M., & Zhou, X. (2024). First-Principles Calculate the Stability, Mechanical Properties and Electronic Structure of Carbide MC, M2C and M6C in M50NiL Steel. Materials, 17(14), 3498. https://doi.org/10.3390/ma17143498