
Impact of PEG Content on Doxorubicin Release from PLGA-co-PEG Nanoparticles

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of PLGA and PLGA-PEG Nanoparticles
2.3. Characterization of NPs
2.3.1. Surface Morphology
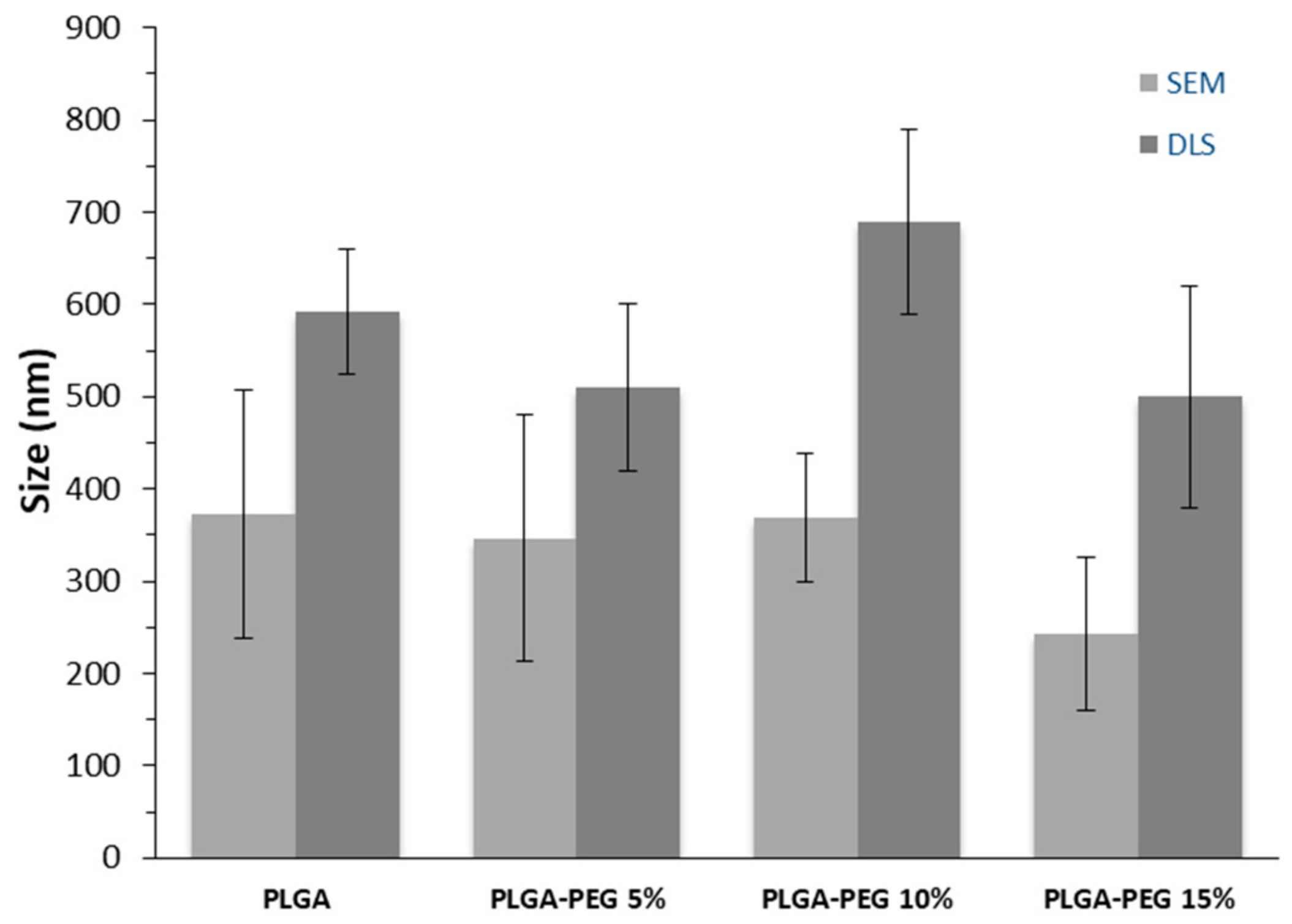
2.3.2. Particles Size and Zeta Potential Analysis
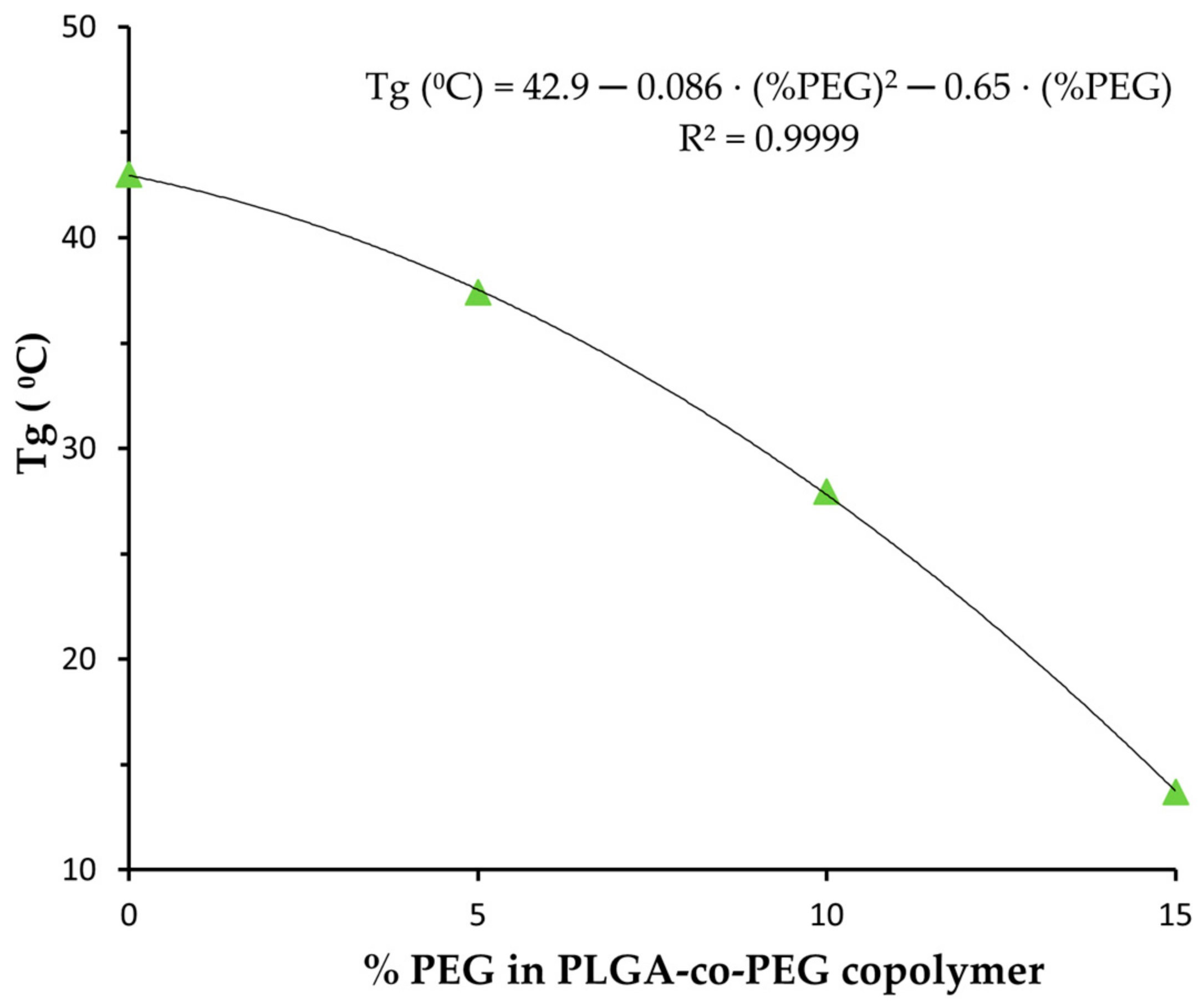
2.3.3. Thermal Analysis by Differential Scanning Calorimetry (DSC)
2.3.4. Drug Loading and Encapsulation Efficiency
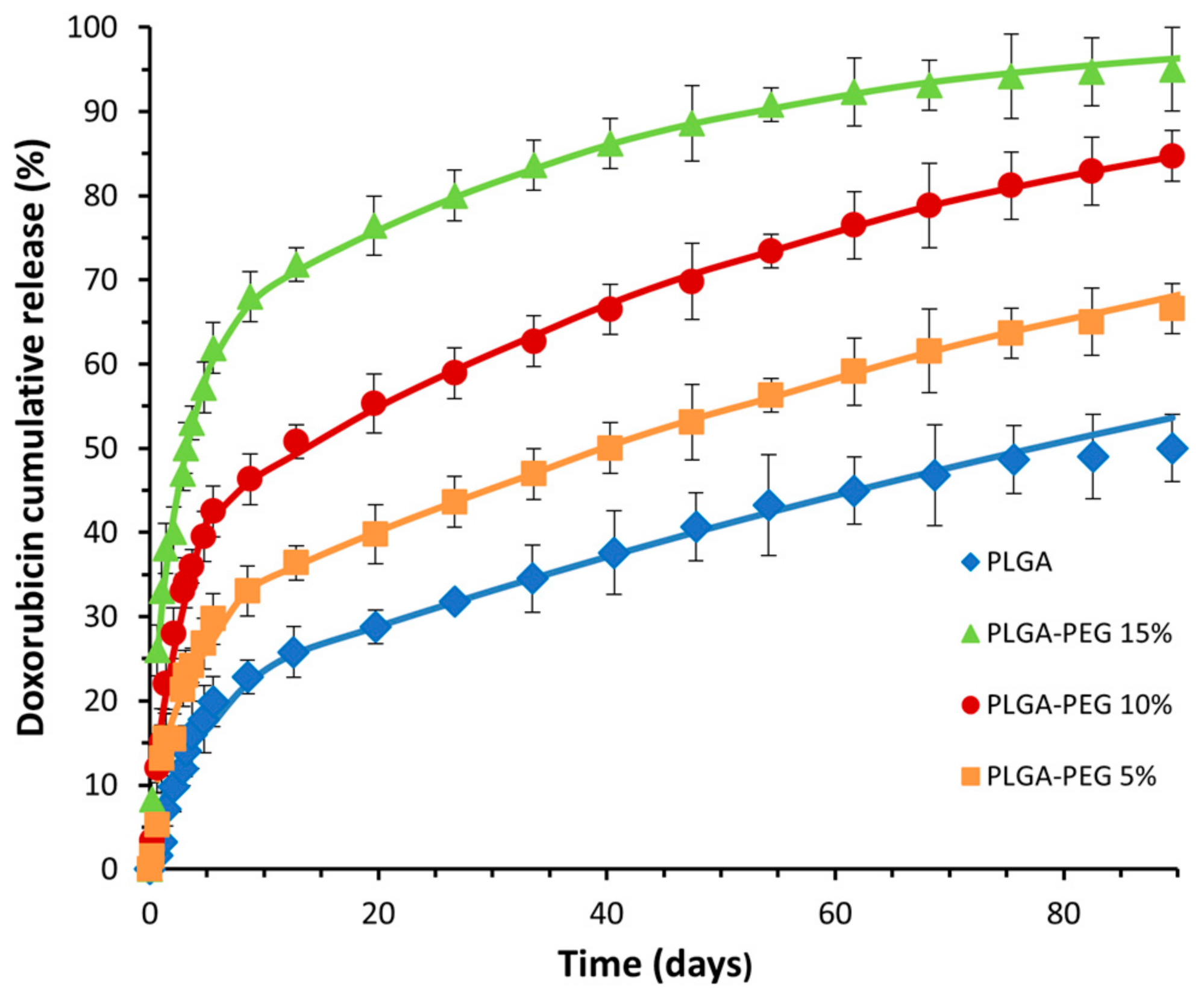
2.4. In Vitro DOX Release Studies
2.5. In Vitro Cell Tests
2.5.1. Cell Cultures
2.5.2. Cell Viability Assays
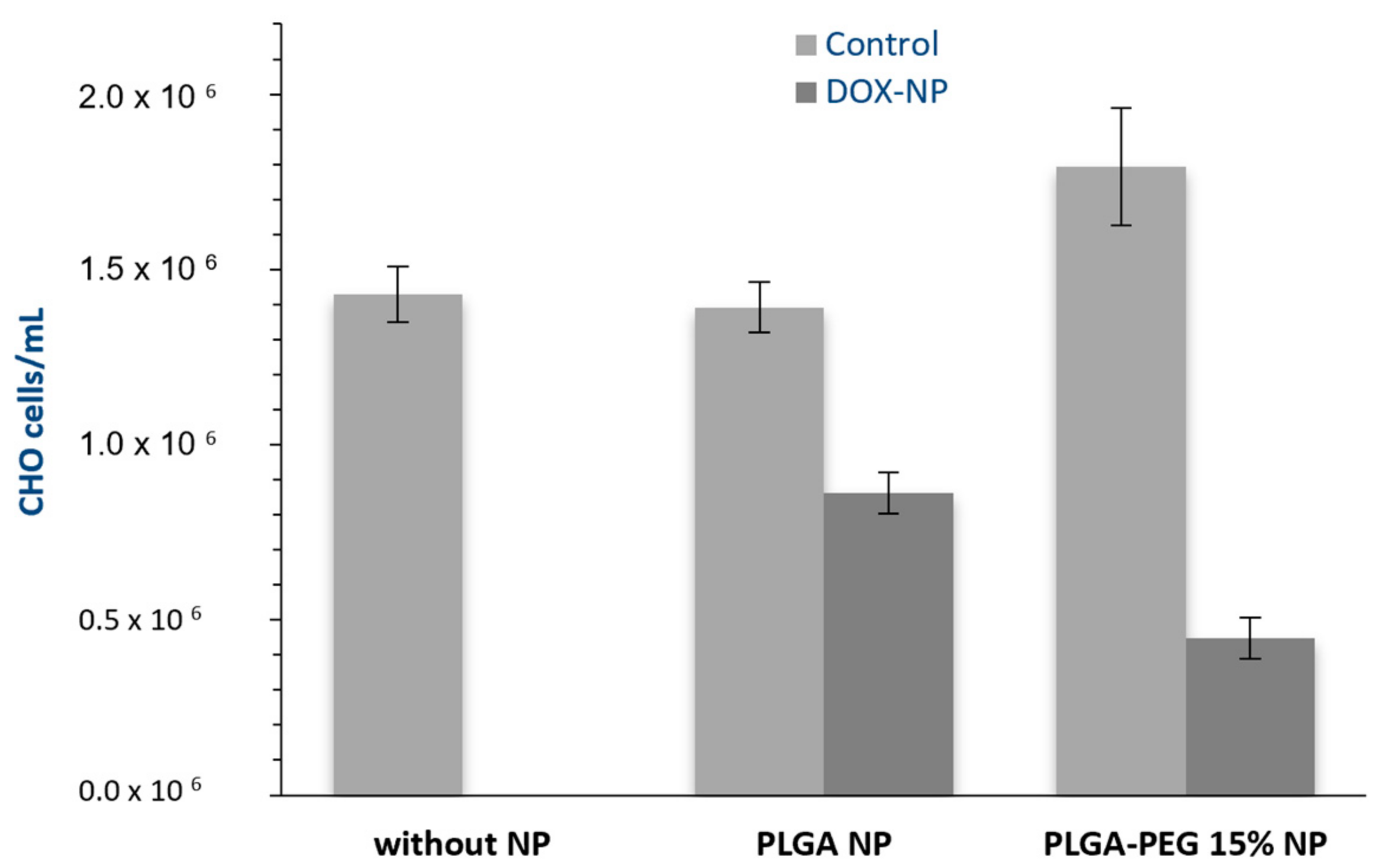
- Absolute Number of Cells
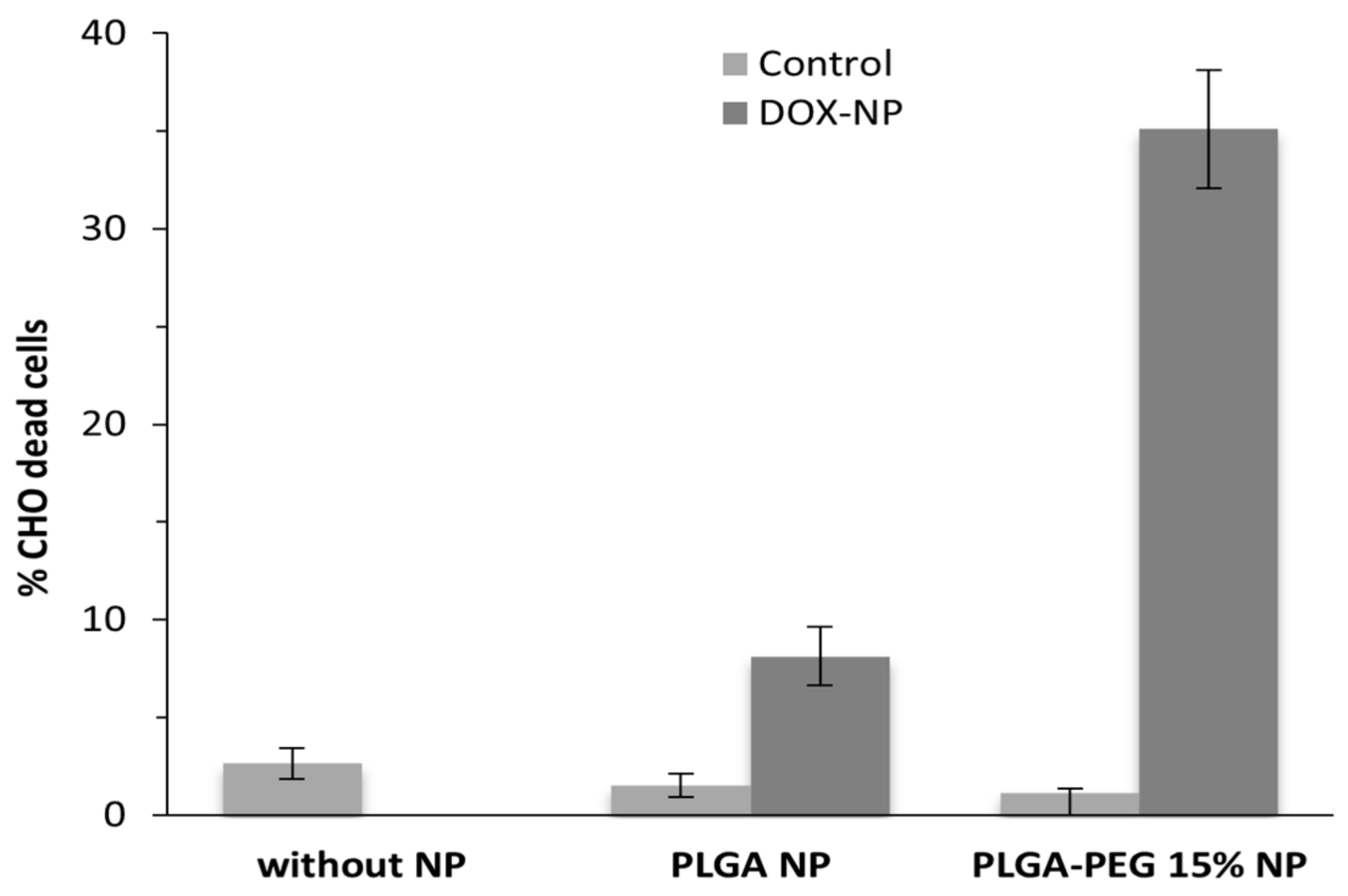
- Flow Cytometry
3. Results and Discussion
3.1. Nanoparticle Characterization
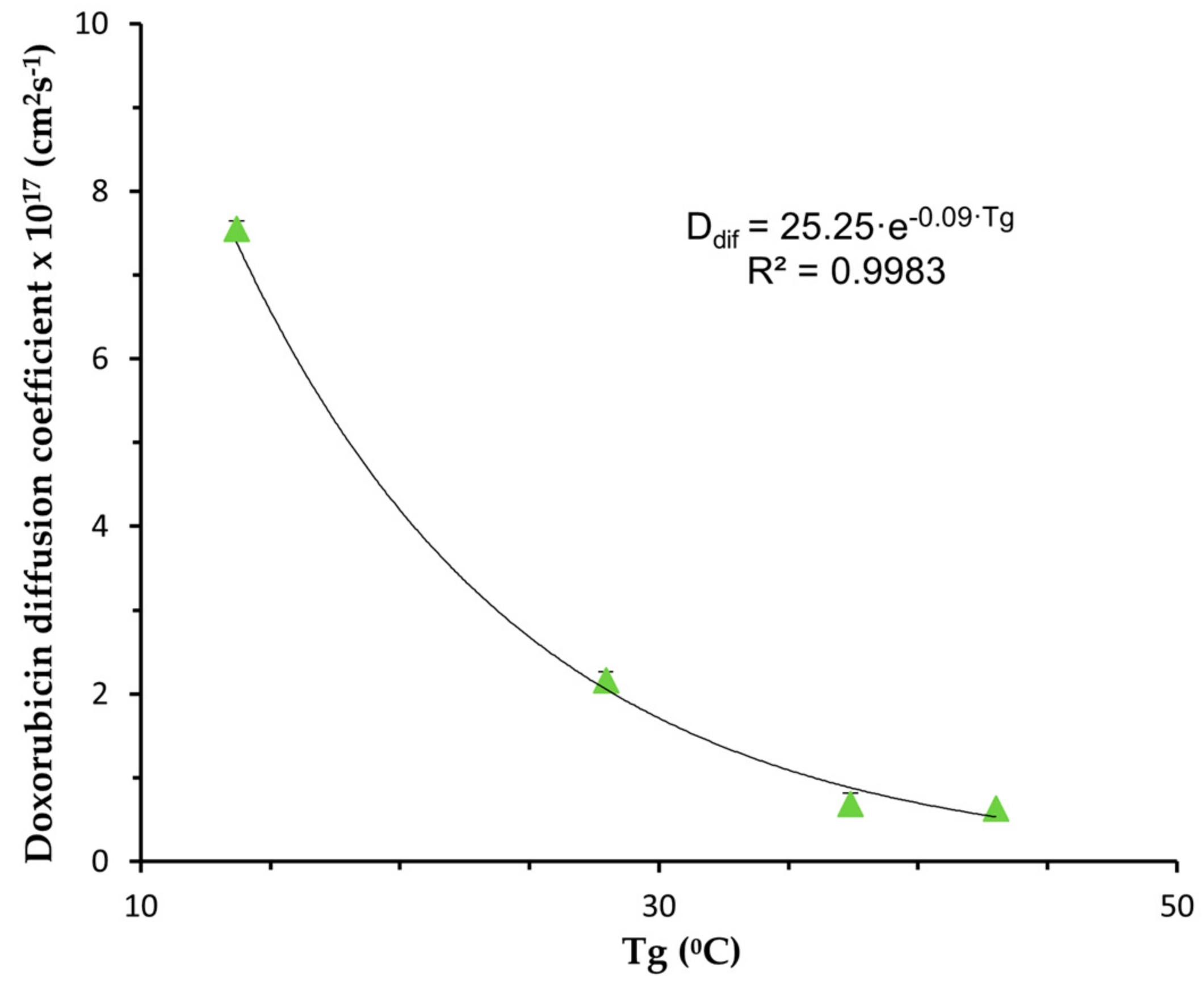
3.2. In Vitro Controlled Drug Release
- Model Fitting
3.3. Viability Assays
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thompson, M. Nanomedicine—A tremendous research opportunity for analytical chemists. Analyst 2004, 129, 671. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Hunter, A.C.; Murray, J.C. Nanomedicine: Current status and future prospects. FASEB J. 2005, 19, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, J. Nanoparticles. In Colloidal Drug Delivery Systems; Kreuter, J., Ed.; Marcel Dekker: New York, NY, USA, 1994. [Google Scholar]
- Cardoso, M.M.; Peça, I.N.; Roque, A.C.A. Antibody-conjugated nanoparticles for therapeutic applications. Curr. Med. Chem. 2012, 19, 3103–3127. [Google Scholar] [CrossRef]
- Arifin, D.Y.; Lee, L.Y.; Wang, C.-H. Mathematical modeling and simulation of drug release from microspheres: Implications to drug delivery systems. Adv. Drug Deliv. Rev. 2006, 58, 1274–1325. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Li, Y.; Kissel, T. Synthesis and properties of biodegradable ABA triblock copolymers consisting of poly (l-lactic acid) or poly (l-lactic-co-glycolic acid) A-blocks attached to central poly (oxyethylene) B-blocks. J. Control. Release 1993, 27, 247–257. [Google Scholar] [CrossRef]
- Li, Y.; Pei, Y.; Zhang, X.; Gu, Z.; Yuan, W.; Zhou, J.; Gao, X. PEGylated PLGA nanoparticles as protein carriers: Synthesis, preparation and biodistribution in rats. J. Control. Release 2001, 71, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Zange, R.; Li, Y.; Kissel, T. Biocompatibility testing of ABA triblock copolymers consisting of poly(L-lactic-co-glycolic acid) A blocks attached to a central poly(ethylene oxide) B block under in vitro conditions using different L929 mouse fibroblasts cell culture models. J. Control. Release 1998, 56, 249–258. [Google Scholar] [CrossRef]
- Davaran, S.; Rashidi, M.R.; Pourabbas, B.; Dadashzadeh, M.; Haghshenas, N.M. Adriamycin release from poly(lactide-coglycolide)-polyethylene glycol nanoparticles: Synthesis, and in vitro characterization. Int. J. Nanomed. 2006, 1, 535–539. [Google Scholar] [CrossRef]
- Wang, H.; Zhao, Y.; Wu, Y.; Hu, Y.-L.; Nan, K.; Nie, G.; Chen, H. Enhanced anti-tumor efficacy by co-delivery of doxorubicin and paclitaxel with amphiphilic methoxy PEG–PLGA copolymer nanoparticles. Biomaterials 2011, 32, 8281–8290. [Google Scholar] [CrossRef]
- Khna, I.; Gothwal, A.; Sharma, A.K.; Qayum, A.; Singh, S.K.; Gupta, U. Biodegradable nano-architectural PEGylated approach for the improved stability and anticancer efficacy of bendamustine. Int. J. Biol. Macromol. 2016, 92, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Mallardé, D.; Boutignon, F.; Moine, F.; Barr, E.; David, S.; Touchet, H.; Ferruti, P.; Deghenghi, R. PLGA-PEG microspheres of teverelix: Influence of polymer type on microsphere characteristics and on teverelix in vitro release. Int. J. Pharm. 2003, 261, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Lochmann, A.; Hagen, N.; von Einem, S.; Schwarz, E.; Mäder, K. The influence of covalently linked and free polyethylene glycol on the structural and release properties of rhBMP-2 loaded microspheres. J. Control. Release 2010, 147, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Yeh, M.-K.; Jenkins, P.G.; Davis, S.S.; Coombes, A.G.A. Improving the delivery capacity of microparticle systems using blends of poly(DL-lactide co-glycolide) and poly(ethyleneglycol). J. Control. Release 1995, 37, 1–9. [Google Scholar] [CrossRef]
- Mäder, K.; Bittner, B.; Li, Y.; Wohlauf, W.; Kissel, T. Monitoring microviscosity and microacidity of the albumin microenvironment inside degrading microparticles from poly (lactide-co-glycolide)(PLG) or ABA-triblock polymers containing hydrophobic poly (lactide-co-glycolide) A blocks and hydrophilic poly (ethyleneoxide) B blocks. Pharm. Res. 1998, 15, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Buske, J.; König, C.; Bassarab, S.; Lamprecht, A.; Mühlau, S.; Wagner, K. Influence of PEG in PEG–PLGA microspheres on particle properties and protein release. Eur. J. Pharm. Biopharm. 2012, 81, 57–63. [Google Scholar] [CrossRef]
- Mollo, A.R.; Corrigan, O.I. Effect of poly-hydroxy aliphatic ester polymer type on amoxycillin release from cylindrical compacts. Int. J. Pharm. 2003, 268, 71–79. [Google Scholar] [CrossRef]
- Corrigan, O.I.; Li, X. Quantifying drug release from PLGA nanoparticulates. Eur. J. Pharm. Sci. 2009, 37, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Arcamone, F. Doxorubicin: Anticancer Antibiotics; Academic Press: New York, NY, USA, 1981. [Google Scholar]
- Bicho, A.; Peça, I.N.; Roque, A.C.A.; Cardoso, M.M. Anti-CD8 conjugated nanoparticles to target mammalian cells expressing CD8. Int. J. Pharm. 2010, 399, 80–86. [Google Scholar] [CrossRef]
- Cardoso, M.M.; Peça, I.N.; Raposo, C.D.; Petrova, K.T.; Barros, M.T.; Gardner, R.; Bicho, A. Doxorubicin-loaded galactose-conjugated poly(d,l-lactide-co-glycolide) nanoparticles as hepatocyte-targeting drug carriers. J. Microencapsul. 2016, 33, 315–322. [Google Scholar] [CrossRef]
- Peça, I.N.; Bicho, A.; Gardner, R.; Cardoso, M.M. Control of doxorubicin release from magnetic poly(dl-lactide-co-glycolide) nanoparticles by application of a non-permanent magnetic field. J. Nanopart. Res. 2015, 17, 427. [Google Scholar] [CrossRef]
- Gupte, A.; Ciftici, K. Formulation and characterization of Paclitaxel, 5-FU and Paclitaxel + 5-FU microspheres. Int. J. Pharm. 2004, 276, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Babos, G.; Biró, E.; Meiczinger, M.; Feczkó, T. Dual Drug Delivery of Sorafenib and Doxorubicin from PLGA and PEG-PLGA Polymeric Nanoparticles. Polymers 2018, 10, 895. [Google Scholar] [CrossRef] [PubMed]
- Silva-Abreu, M.; Espinoza, L.C.; Halbaut, L.; Espina, M.; García, M.L.; Calpena, A.C. Comparative Study of Ex Vivo Transmucosal Permeation of Pioglitazone Nanoparticles for the Treatment of Alzheimer’s Disease. Polymers 2018, 10, 316. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Feng, S.S. A novel controlled release formulation for the anticancer drug paclitaxel (Taxol®): PLGA nanoparticles containing vitamin E TPGS. J. Control. Release 2003, 86, 33–48. [Google Scholar] [CrossRef]
- Jeong, B.; Baeb, Y.H.; Kim, S.W. Drug release from biodegradable injectable thermosensitive hydrogel of PEG–PLGA–PEG triblock copolymers. J. Control. Release 2000, 63, 155–163. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NP | Zeta Potential (mV) | DOX Loading (mg DOX/100 mg NP) | EE (%) | Tg (°C) |
|---|---|---|---|---|
| PLGA | −14.4 ± 0.8 | 2.9 ± 0.6 | 18 ± 4 | 43.1 |
| PLGA-PEG 5% | −9.8 ± 0.7 | 2.8 ± 0.8 | 187 ± 5 | 37.4 |
| PLGA-PEG 10% | −6.5 ± 0.7 | 2.6 ± 0.6 | 16 ± 4 | 27.9 |
| PLGA-PEG 15% | −9.9 ± 0.9 | 2.8 ± 0.7 | 17 ± 4 | 13.7 |
| NP | Diffusion Coefficient (cm2s−1) | R2adj | MSC |
|---|---|---|---|
| PLGA | (6.30 ± 0.13) × 10−18 | 0.9952 | 5.22 |
| PLGA-PEG 5% | (6.88 ± 0.08) × 10−18 | 0.9999 | 6.62 |
| PLGA-PEG 10% | (2.16 ± 0.03) × 10−17 | 0.9999 | 5.95 |
| PLGA-PEG 15% | (7.55 ± 0.16) × 10−17 | 0.9989 | 4.52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardoso, M.M.; Peça, I.N.; Bicho, A. Impact of PEG Content on Doxorubicin Release from PLGA-co-PEG Nanoparticles. Materials 2024, 17, 3544. https://doi.org/10.3390/ma17143544
Cardoso MM, Peça IN, Bicho A. Impact of PEG Content on Doxorubicin Release from PLGA-co-PEG Nanoparticles. Materials. 2024; 17(14):3544. https://doi.org/10.3390/ma17143544
Chicago/Turabian StyleCardoso, Maria Margarida, Inês N. Peça, and Ana Bicho. 2024. "Impact of PEG Content on Doxorubicin Release from PLGA-co-PEG Nanoparticles" Materials 17, no. 14: 3544. https://doi.org/10.3390/ma17143544