

Configurational Isomerism in Bimetallic Decametalates


Abstract

1. Introduction
2. Materials and Methods
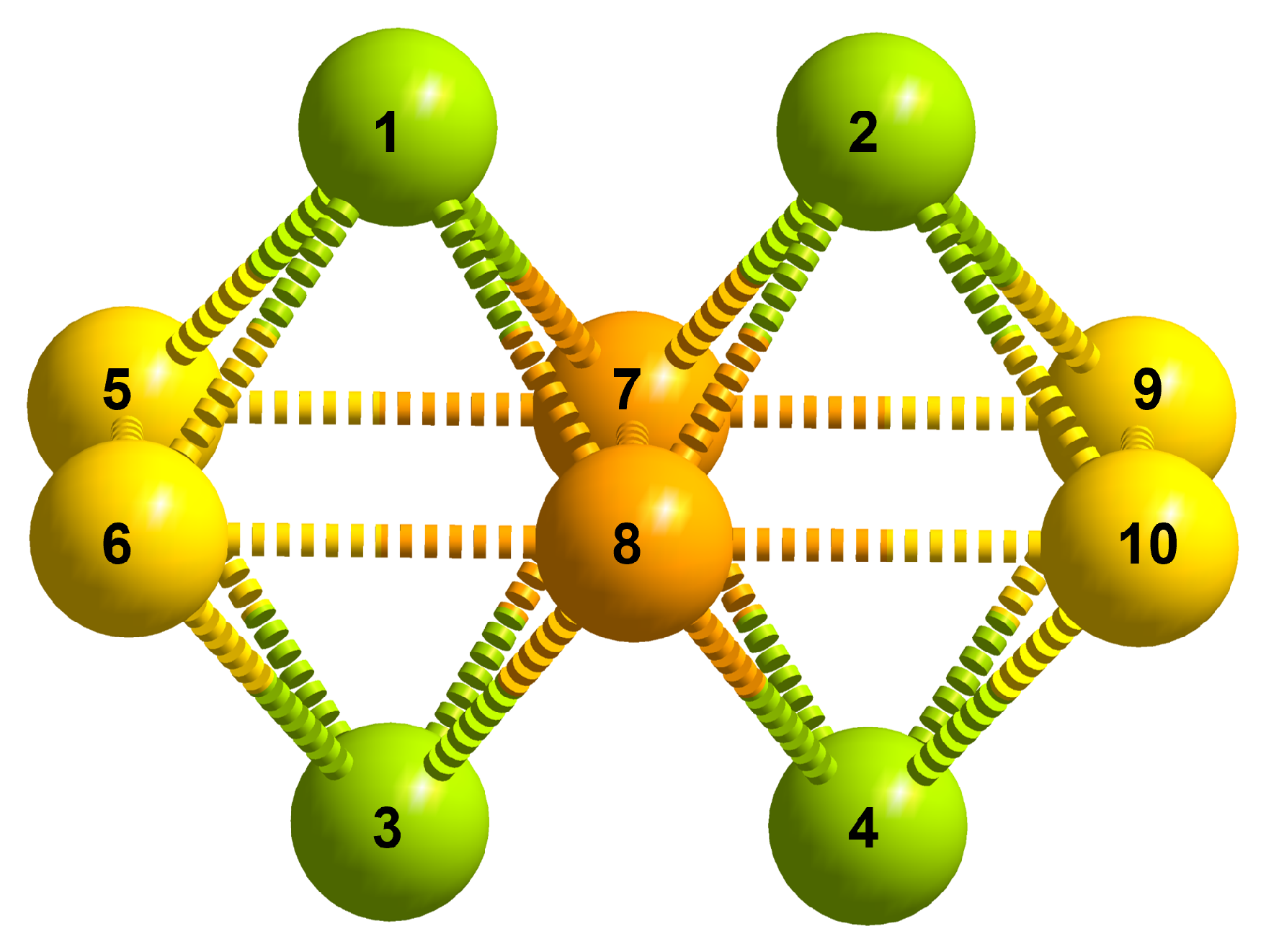
2.1. Decametalate Isomerizer
| Algorithm 1: Decametalate Isomerizer |
| Input: Number of positions n Output: Set of all unique configurations  |
2.2. Molecular DFT Calculations
3. Results
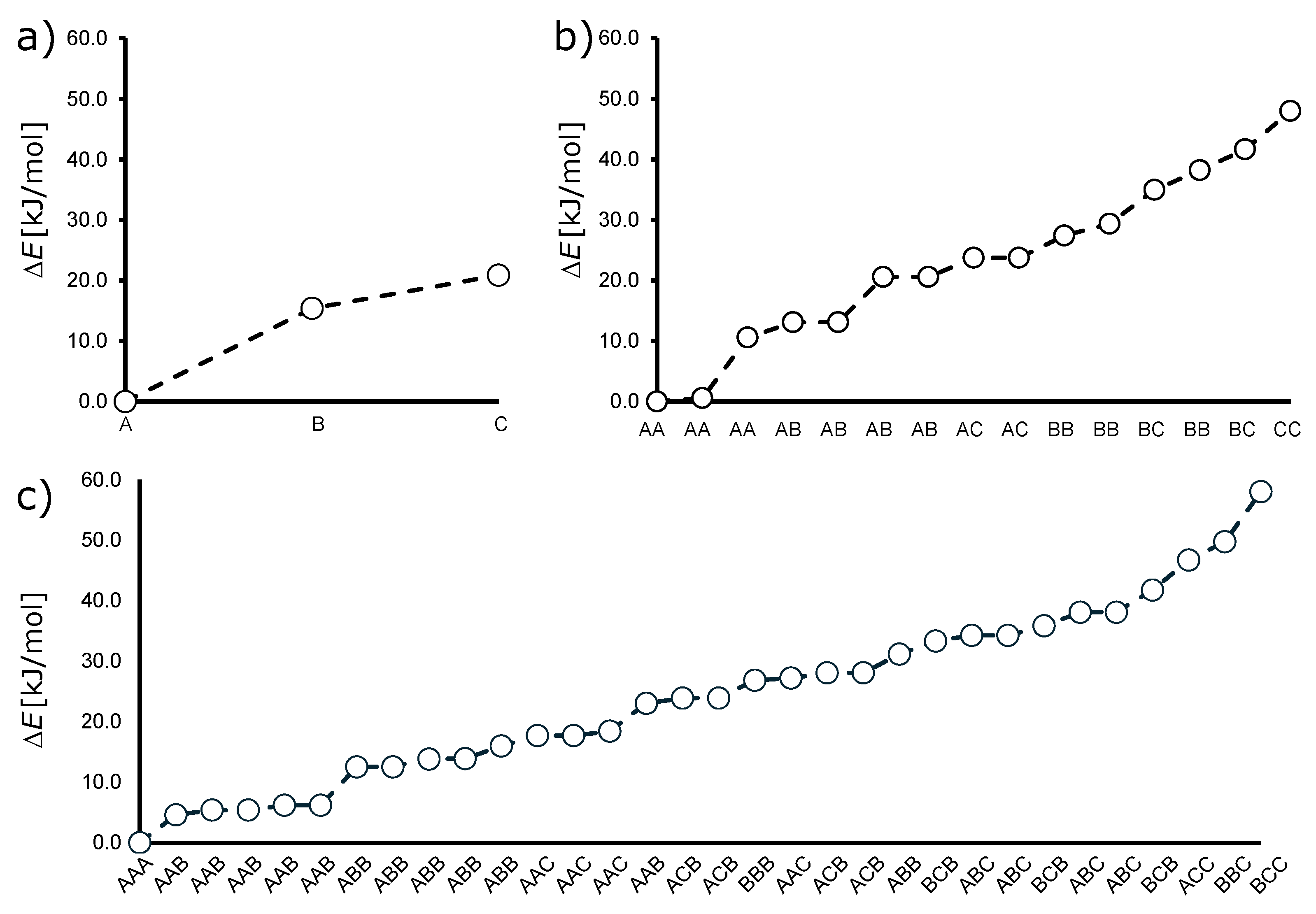
3.1. Total Number of Isomers
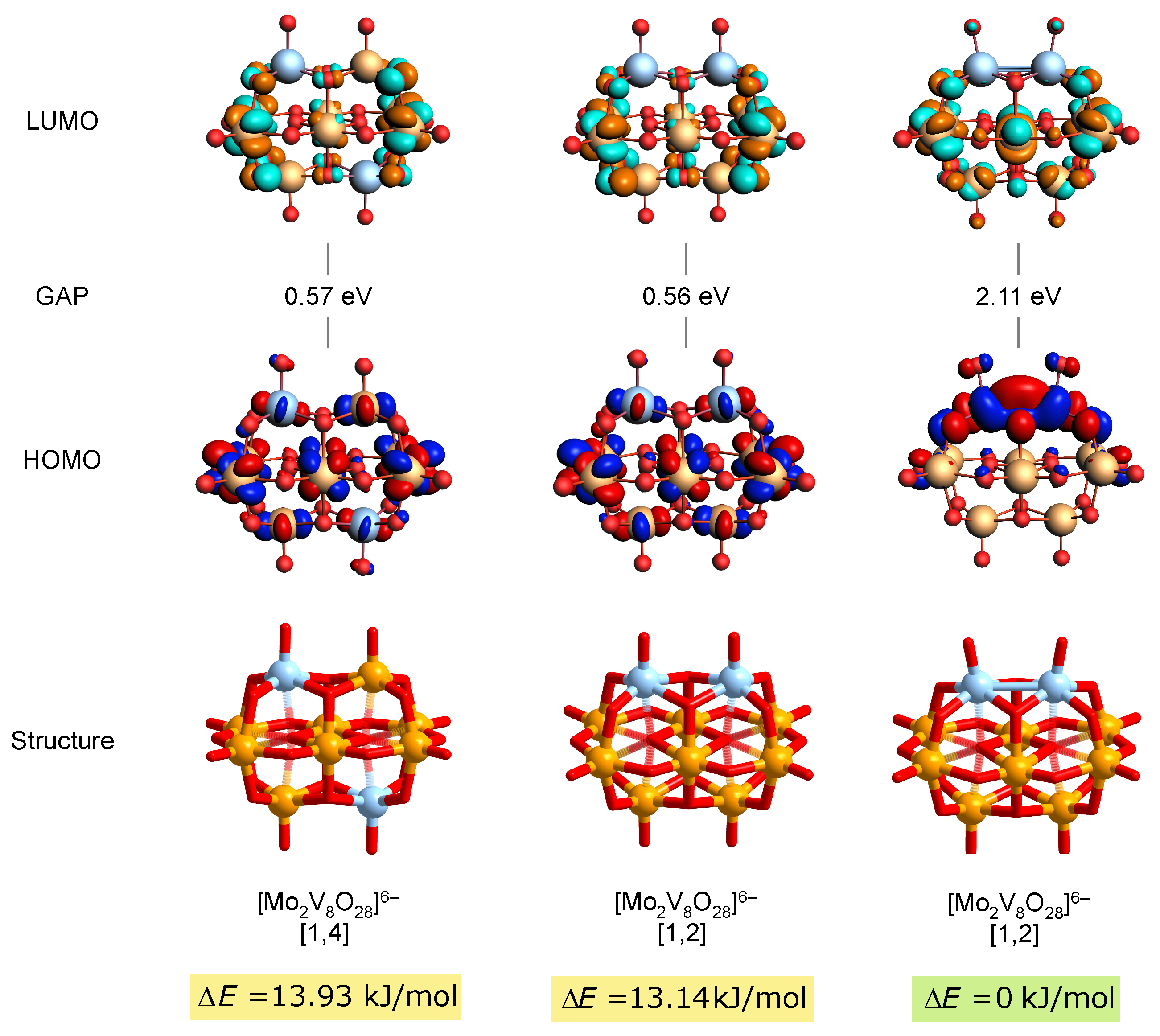
3.2. Structure, Relative Stability, and Electronic Properties
3.3. Two-Electron Reduction and Metal–Metal Bonding
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| POMs | Polyoxometalates |
| TZP | Triple-Zeta Quality with Polarization (basis set in quantum chemistry) |
| ZORA | Zero-Order Regular Approximation (a relativistic treatment method) |
| COSMO | Conductor-like Screening Model (a method for including solvent effects in simulations) |
| BP | Becke exchange plus the Perdew 86 correlation functional (a type of density functional) |
| B3LYP | Becke, three-parameter, Lee–Yang–Parr (a hybrid functional in density functional theory) |
| DFT | Density Functional Theory |
| GGA | Generalized Gradient Approximation (a type of exchange-correlation functional in DFT) |
| HOMO | Highest Occupied Molecular Orbital |
| LUMO | Lowest Unoccupied Molecular Orbital |
| NMR | Nuclear Magnetic Resonance |
References
- Liu, R.; Streb, C. Polyoxometalate-single atom catalysts (POM-SACs) in energy research and catalysis. Adv. Energy Mater. 2021, 11, 2101120. [Google Scholar] [CrossRef]
- Gumerova, N.I.; Rompel, A. Interweaving disciplines to advance chemistry: Applying polyoxometalates in biology. Inorg. Chem. 2021, 60, 6109–6114. [Google Scholar] [CrossRef]
- Barba-Bon, A.; Gumerova, N.I.; Tanuhadi, E.; Ashjari, M.; Chen, Y.; Rompel, A.; Nau, W.M. All-Inorganic Polyoxometalates Act as Superchaotropic Membrane Carriers. Adv. Mater. 2024, 36, 2309219. [Google Scholar] [CrossRef]
- Kondinski, A. Metal–metal bonds in polyoxometalate chemistry. Nanoscale 2021, 13, 13574–13592. [Google Scholar] [CrossRef]
- Kondinski, A.; Banerjee, A.; Mal, S.S. Hervé- and Krebs-Type Magnetic Polyoxometalate Dimers. Magnetochemistry 2022, 8, 96. [Google Scholar] [CrossRef]
- Kondinski, A.; Monakhov, K.Y. Breaking the Gordian knot in the structural chemistry of polyoxometalates: Copper (II)–oxo/hydroxo clusters. Chem. Eur. J. 2017, 23, 7841–7852. [Google Scholar] [CrossRef]
- Nyman, M.; Burns, P.C. A comprehensive comparison of transition-metal and actinyl polyoxometalates. Chem. Soc. Rev. 2012, 41, 7354–7367. [Google Scholar] [CrossRef]
- Kondinski, A.; Parac-Vogt, T.N. Keggin structure, quō vādis? Front. Chem. 2018, 6, 346. [Google Scholar] [CrossRef]
- Linnenberg, O.; Kondinski, A.; Monakhov, K.Y. The Lindqvist hexavanadate: A platform for coordination-directed assembly. In Supramolecular Systems: Chemistry, Types and Applications; Nova Science Publishers: Hauppauge, NY, USA, 2017; pp. 39–66. [Google Scholar]
- Blazevic, A.; Rompel, A. The Anderson–Evans polyoxometalate: From inorganic building blocks via hybrid organic–inorganic structures to tomorrows “Bio-POM”. Coord. Chem. Rev. 2016, 307, 42–64. [Google Scholar] [CrossRef]
- Mirzaei, M.; Eshtiagh-Hosseini, H.; Alipour, M.; Frontera, A. Recent developments in the crystal engineering of diverse coordination modes (0–12) for Keggin-type polyoxometalates in hybrid inorganic–organic architectures. Coord. Chem. Rev. 2014, 275, 1–18. [Google Scholar] [CrossRef]
- Ghosh, M.; Sorsche, D.; Ahmed, R.B.; Anjass, M. Stabilizing Decavanadate Cluster as Electrode Material in Sodium and Lithium-ion Batteries. ChemSusChem 2023, 16, e202300631. [Google Scholar] [CrossRef]
- Kruse, J.H.; Langer, M.; Romanenko, I.; Trentin, I.; Hernández-Castillo, D.; González, L.; Schacher, F.H.; Streb, C. Polyoxometalate-soft matter composite materials: Design strategies, applications, and future directions. Adv. Funct. Mater. 2022, 32, 2208428. [Google Scholar] [CrossRef]
- Kikukawa, Y.; Ogihara, K.; Hayashi, Y. Structure Transformation among Deca-, Dodeca- and Tridecavanadates and Their Properties for Thioanisole Oxidation. Inorganics 2015, 3, 295–308. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, Y.; Li, G.; Wei, Y. Recent advances in alkoxylation chemistry of polyoxometalates: From synthetic strategies, structural overviews to functional applications. Coord. Chem. Rev. 2019, 378, 395–414. [Google Scholar] [CrossRef]
- Weinstock, I.A.; Schreiber, R.E.; Neumann, R. Dioxygen in polyoxometalate mediated reactions. Chem. Rev. 2017, 118, 2680–2717. [Google Scholar] [CrossRef]
- Simms, C.; Kondinski, A.; Parac-Vogt, T.N. Metal-Addenda Substitution in Plenary Polyoxometalates and in Their Modular Transition Metal Analogues. Eur. J. Inorg. Chem. 2020, 2020, 2559–2572. [Google Scholar] [CrossRef]
- Kondinski, A. Computational modelling of isomeric polyoxometalates. Chem. Model. 2021, 16, 39–71. [Google Scholar]
- Khenkin, A.M.; Rosenberger, A.; Neumann, R. Reaction of Aldehydes with the H5PV2Mo10O40 Polyoxometalate and Cooxidation of Alkanes with Molecular Oxygen. J. Catal. 1999, 182, 82–91. [Google Scholar] [CrossRef]
- Wu, S.; Zhao, C.; Dong, Y.; Yan, L. Cyclohexanone hydrogenation to cyclohexanol on phosphomolybdate supported Pt single-atom catalyst: A density functional theory study. Polyoxometalates 2024, 3, 9140070. [Google Scholar] [CrossRef]
- Vongtragool, S.; Gorshunov, B.; Mukhin, A.A.; van Slageren, J.; Dressel, M.; Müller, A. High-frequency magnetic spectroscopy on the molecular magnetic cluster V 15. Phys. Chem. Chem. Phys. 2003, 5, 2778–2782. [Google Scholar] [CrossRef]
- Mahnke, L.K.; Kondinski, A.; Warzok, U.; Näther, C.; Van Leusen, J.; Schalley, C.A.; Monakhov, K.Y.; Kögerler, P.; Bensch, W. Configurational isomerism in polyoxovanadates. Angew. Chem., Int. Ed. 2018, 57, 2972–2975. [Google Scholar] [CrossRef]
- Izarova, N.V.; Kondinski, A.; Vankova, N.; Heine, T.; Jäger, P.; Schinle, F.; Hampe, O.; Kortz, U. The Mixed Gold–Palladium Polyoxo-Noble-Metalate [NaAuIII4PdII8O8(AsO4)8]11-. Chem. Eur. J. 2014, 20, 8556–8560. [Google Scholar] [CrossRef]
- Kondinski, A.; Heine, T.; Monakhov, K.Y. Rotational Isomerism, Electronic Structures, and Basicity Properties of “Fully-Reduced” V14-type Heteropolyoxovanadates. Inorg. Chem. 2016, 55, 3777–3788. [Google Scholar] [CrossRef]
- Pope, M.T.; Scully, T.F. Geometrical isomerism arising from partial substitution of metal atoms in isopoly and heteropoly complexes. Possibilities for the Keggin structure. Inorg. Chem. 1975, 14, 953–954. [Google Scholar] [CrossRef]
- López, X.; Carbó, J.J.; Bo, C.; Poblet, J.M. Structure, properties and reactivity of polyoxometalates: A theoretical perspective. Chem. Soc. Rev. 2012, 41, 7537–7571. [Google Scholar] [CrossRef]
- Kondinski, A.; Rasmussen, M.; Mangelsen, S.; Pienack, N.; Simjanoski, V.; Näther, C.; Stares, D.L.; Schalley, C.A.; Bensch, W. Composition-driven archetype dynamics in polyoxovanadates. Chem. Sci. 2022, 13, 6397–6412. [Google Scholar] [CrossRef]
- Dumitrescu, A.; Maxim, C.; Badea, M.; Rostas, A.M.; Ciorîță, A.; Tirsoaga, A.; Olar, R. Decavanadate-Bearing Guanidine Derivatives Developed as Antimicrobial and Antitumor Species. Int. J. Mol. Sci. 2023, 24, 17137. [Google Scholar] [CrossRef]
- Aminifazl, A.; Karunarathne, D.J.; Golden, T.D. Synthesis of Silane Functionalized LDH-Modified Nanopowders to Improve Compatibility and Enhance Corrosion Protection for Epoxy Coatings. Molecules 2024, 29, 819. [Google Scholar] [CrossRef]
- Nyman, M. Polyoxoniobate chemistry in the 21st century. Dalton Trans. 2011, 40, 8049–8058. [Google Scholar] [CrossRef]
- Aureliano, M.; Ohlin, C.A.; Vieira, M.O.; Marques, M.P.M.; Casey, W.H.; De Carvalho, L.A.B. Characterization of decavanadate and decaniobate solutions by Raman spectroscopy. Dalton Trans. 2016, 45, 7391–7399. [Google Scholar] [CrossRef]
- Matsumoto, M.; Ozawa, Y.; Yagasaki, A.; Zhe, Y. Decatantalate?The Last Member of the Group 5 Decametalate Family. Inorg. Chem. 2013, 52, 7825–7827. [Google Scholar] [CrossRef]
- Graeber, E.J.; Morosin, B. The molecular configuration of the decaniobate ion (Nb10O286-). Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1977, 33, 2137–2143. [Google Scholar] [CrossRef]
- Lee, U.; Joo, H.C.; Park, K.M.; Mal, S.S.; Kortz, U.; Keita, B.; Nadjo, L. Facile incorporation of platinum (IV) into polyoxometalate frameworks: Preparation of [H2PtIVV9O28] 5- and characterization by 195Pt NMR spectroscopy. Angew. Chem. Int. Ed. 2008, 47, 793–796. [Google Scholar] [CrossRef]
- Joo, H.C.; Park, K.M.; Lee, U. Double salt crystal structure of nonasodium dihydrogen nonavanadoplatinate (IV) trihydrogen nonavanadoplatinate (IV) tetracontahydrate: Stepwise-protonated nonavanadoplatinate (IV). Acta Crystallogr. Sect. E Crystallogr. Commun. 2015, 71, 786–790. [Google Scholar] [CrossRef]
- Son, J.H.; Ohlin, C.A.; Casey, W.H. Highly soluble iron-and nickel-substituted decaniobates with tetramethylammonium countercations. Dalton Trans. 2013, 42, 7529–7533. [Google Scholar] [CrossRef]
- Ohlin, C.A.; Villa, E.M.; Fettinger, J.C.; Casey, W.H. A new titanoniobate ion—Completing the series [Nb10O28]6-, [TiNb9O28]7- and [Ti2Nb8O28]8-. Dalton Trans. 2009, 2677–2678. [Google Scholar] [CrossRef] [PubMed]
- Fukui, A.; Ozawa, Y.; Yagasaki, A. Synthesis and structure of vanadoperiodate. Polyhedron 2012, 42, 149–152. [Google Scholar] [CrossRef]
- Konaka, S.; Ozawa, Y.; Shonaka, T.; Watanabe, S.; Yagasaki, A. [HxTeV9O28](5-x)- (x = 1 and 2): Vanadotellurates with Decavanadate Structure. Inorg. Chem. 2011, 50, 6183–6188. [Google Scholar] [CrossRef]
- Spillane, S.; Sharma, R.; Zavras, A.; Mulder, R.; Ohlin, C.A.; Goerigk, L.; O’Hair, R.A.; Ritchie, C. Non-Aqueous Microwave-Assisted Syntheses of Deca-and Hexa-Molybdovanadates. Angew. Chem. Int. Ed. 2017, 56, 8568–8572. [Google Scholar] [CrossRef]
- Howarth, O.W.; Pettersson, L.; Andersson, I. Monomolybdononavanadate and cis-and trans-dimolybdo-octavanadate. J. Chem. Soc. Dalton Trans. 1989, 1915–1923. [Google Scholar] [CrossRef]
- Nyman, M.; Criscenti, L.J.; Bonhomme, F.; Rodriguez, M.A.; Cygan, R.T. Synthesis, structure, and molecular modeling of a titanoniobate isopolyanion. J. Solid State Chem. 2003, 176, 111–119. [Google Scholar] [CrossRef]
- Te Velde, G.t.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Amsterdam Density Functional (ADF). Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands, 2022. [Google Scholar]
- Van Gisbergen, S.; Snijders, J.; Baerends, E. Implementation of time-dependent density functional response equations. Comput. Phys. Commun. 1999, 118, 119–138. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1–118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A. Conductor-like screening model for real solvents: A new approach to the quantitative calculation of solvation phenomena. J. Phys. Chem. 1995, 99, 2224–2235. [Google Scholar] [CrossRef]
- Pye, C.C.; Ziegler, T. An implementation of the conductor-like screening model of solvation within the Amsterdam density functional package. Theor. Chem. Acc. 1999, 101, 396–408. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Ehlers, A.; Baerends, E.J. Geometry optimizations in the zero-order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef]
- Avila, P.F.; Ripplinger, T.J.; Kemper, D.J.; Domine, J.L.; Jordan, C.D. Features of Vibrational and Electronic Structures of Decavanadate Revealed by Resonance Raman Spectroscopy and Density Functional Theory. J. Phys. Chem. Lett. 2019, 10, 6032–6037. [Google Scholar] [CrossRef]
- Cao, M.; Zi, J.; Sang, R.; Xu, L. Metal–metal bonded pentamolybdate hybrids as electron storage materials. Dalton Trans. 2023, 52, 13351–13357. [Google Scholar] [CrossRef]
- Gumerova, N.I.; Rompel, A. Synthesis, structures and applications of electron-rich polyoxometalates. Nat. Rev. Chem. 2018, 2, 0112. [Google Scholar] [CrossRef]
- Lang, Z.; Gabas, I.M.; López, X.; Clotet, A.; de la Fuente, J.M.; Mitchell, S.G.; Poblet, J.M. On the formation of gold nanoparticles from [AuIIICl4]- and a non-classical reduced polyoxomolybdate as an electron source: A quantum mechanical modelling and experimental study. New J. Chem. 2016, 40, 1029–1038. [Google Scholar] [CrossRef]
- Rohmer, M.M.; Bénard, M. Metastable states associated with a change in the metal–metal bonding network of (MoV)6 polyoxoanions: A DFT study of [(Mo2VO4)3 (μ6-CO3)(μ-CO3)3 (μ-OH)3]5-. Dalton Trans. 2003, 3587–3590. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| x | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| unique configomers | 1 | 3 | 15 | 32 | 60 | 66 | 60 | 32 | 15 | 3 | 1 |
| enantiomeric pairs | 0 | 0 | 3 | 9 | 19 | 21 | 19 | 9 | 3 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kondinski, A. Configurational Isomerism in Bimetallic Decametalates. Materials 2024, 17, 3624. https://doi.org/10.3390/ma17143624
Kondinski A. Configurational Isomerism in Bimetallic Decametalates. Materials. 2024; 17(14):3624. https://doi.org/10.3390/ma17143624
Chicago/Turabian StyleKondinski, Aleksandar. 2024. "Configurational Isomerism in Bimetallic Decametalates" Materials 17, no. 14: 3624. https://doi.org/10.3390/ma17143624
APA StyleKondinski, A. (2024). Configurational Isomerism in Bimetallic Decametalates. Materials, 17(14), 3624. https://doi.org/10.3390/ma17143624