
Solid Dispersions Obtained by Ball Milling as Delivery Platform of Etodolac, a Model Poorly Soluble Drug
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. High-Performance Liquid Chromatography Analysis
2.3. Preliminary Studies—Development of the Preparation Method, Formulation, and Characterization of the 1st and 2nd Generation of ETD-SDs
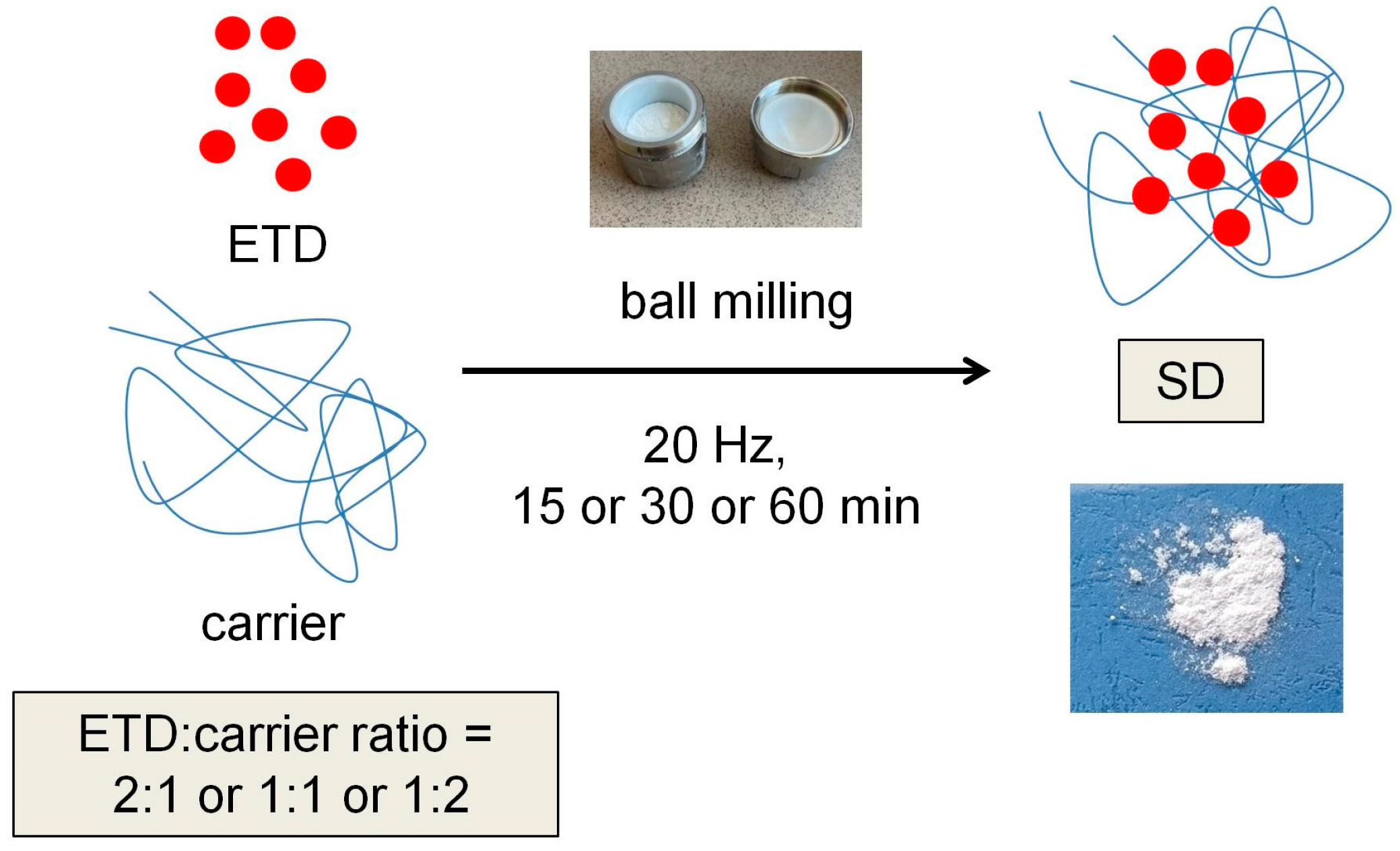
2.3.1. Preparation of ETD-SDs
2.3.2. Solubility Studies
2.3.3. Differential Scanning Calorimetry (DSC)
2.4. Solid Dispersion with ETD—2nd and 3rd Generation
2.4.1. Preparation of ETD-SDs
2.4.2. Drug Content Studies
2.4.3. Solubility Analysis and Dissolution Testing
2.4.4. Morphological Assessment
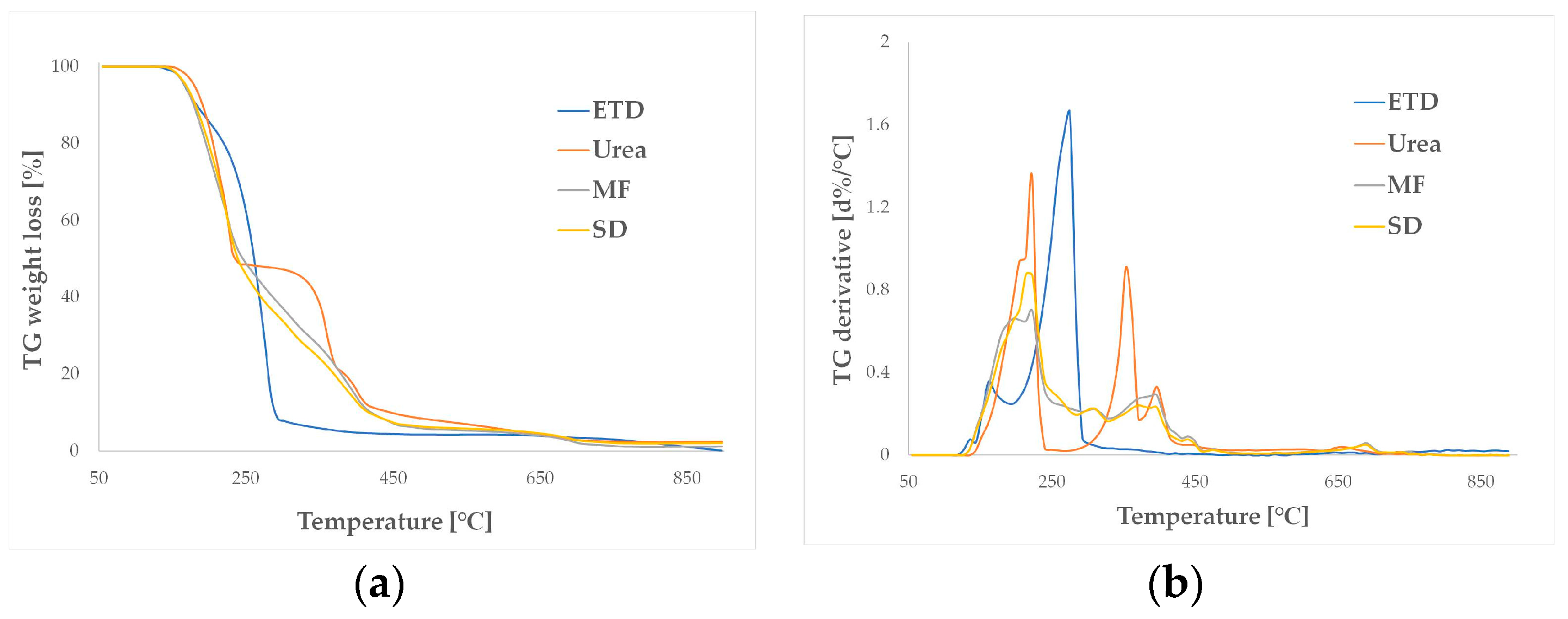
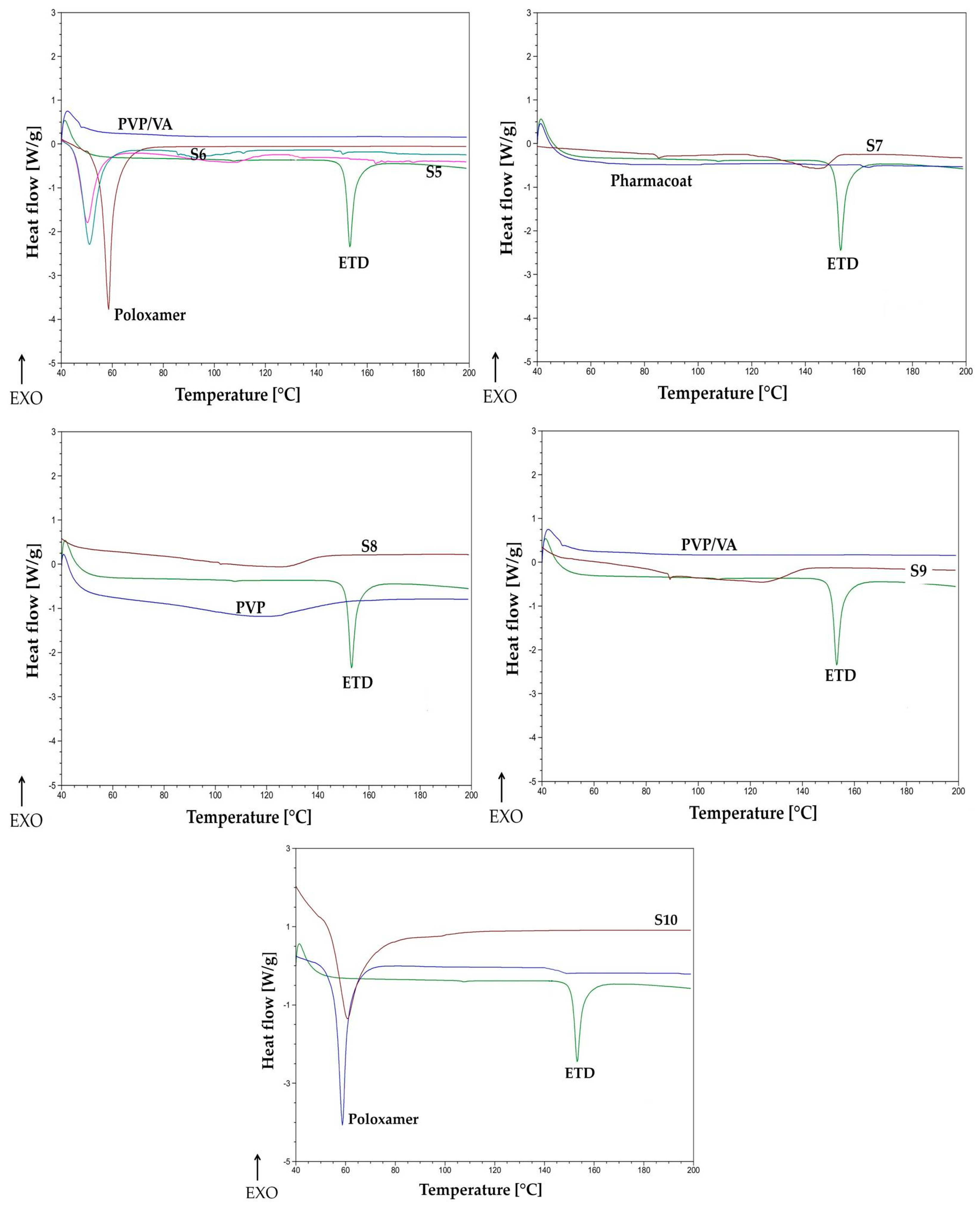
2.4.5. Thermal Analysis
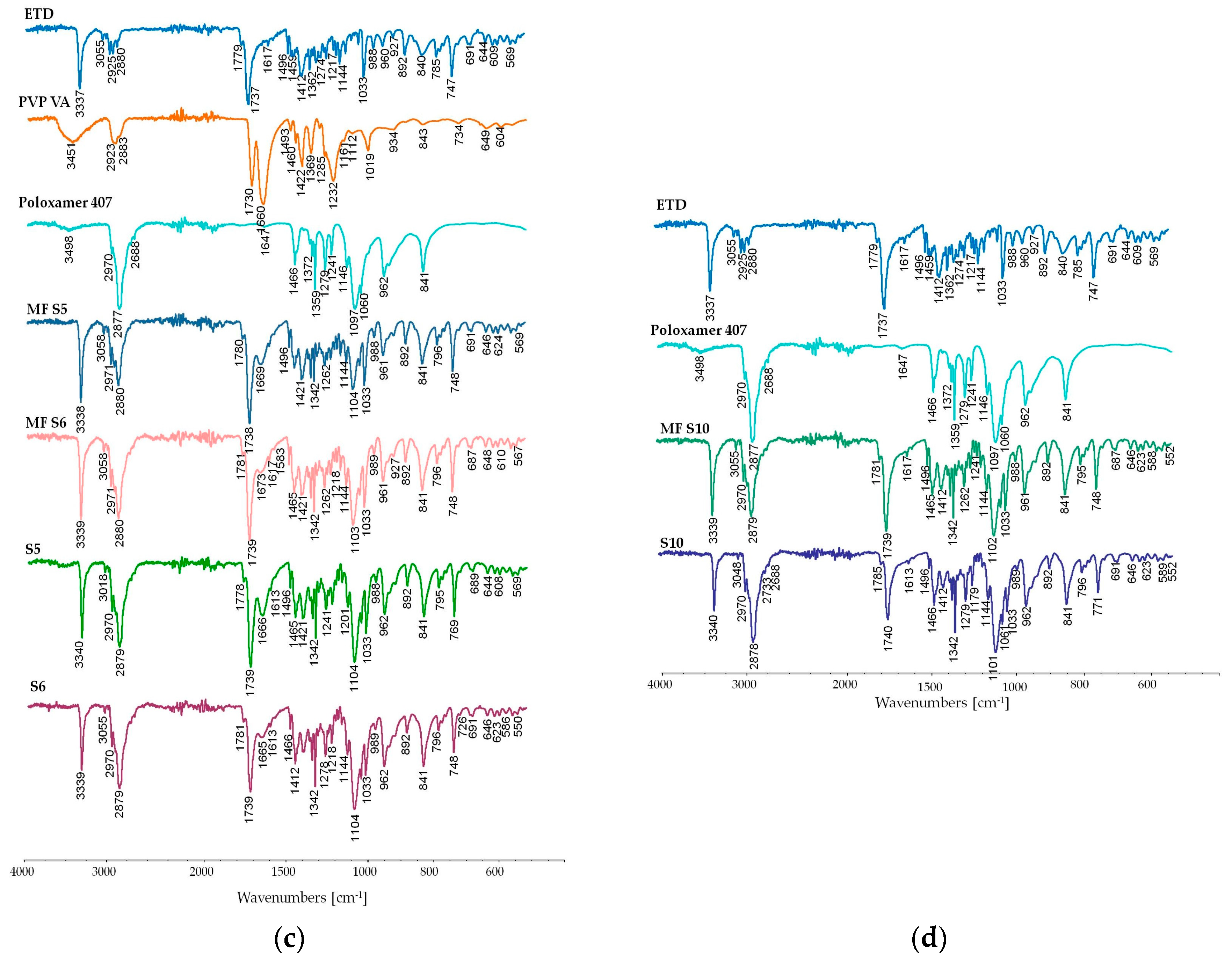
2.4.6. Attenuated Total Reflectance Fourier Transform Infrared Spectroscopy (ATR-FTIR)
2.5. Statistical Analysis
3. Results and Discussion
3.1. Preliminary Studies—1st and 2nd Generation of ETD-SDs
3.1.1. Solubility
3.1.2. Thermal Evaluation
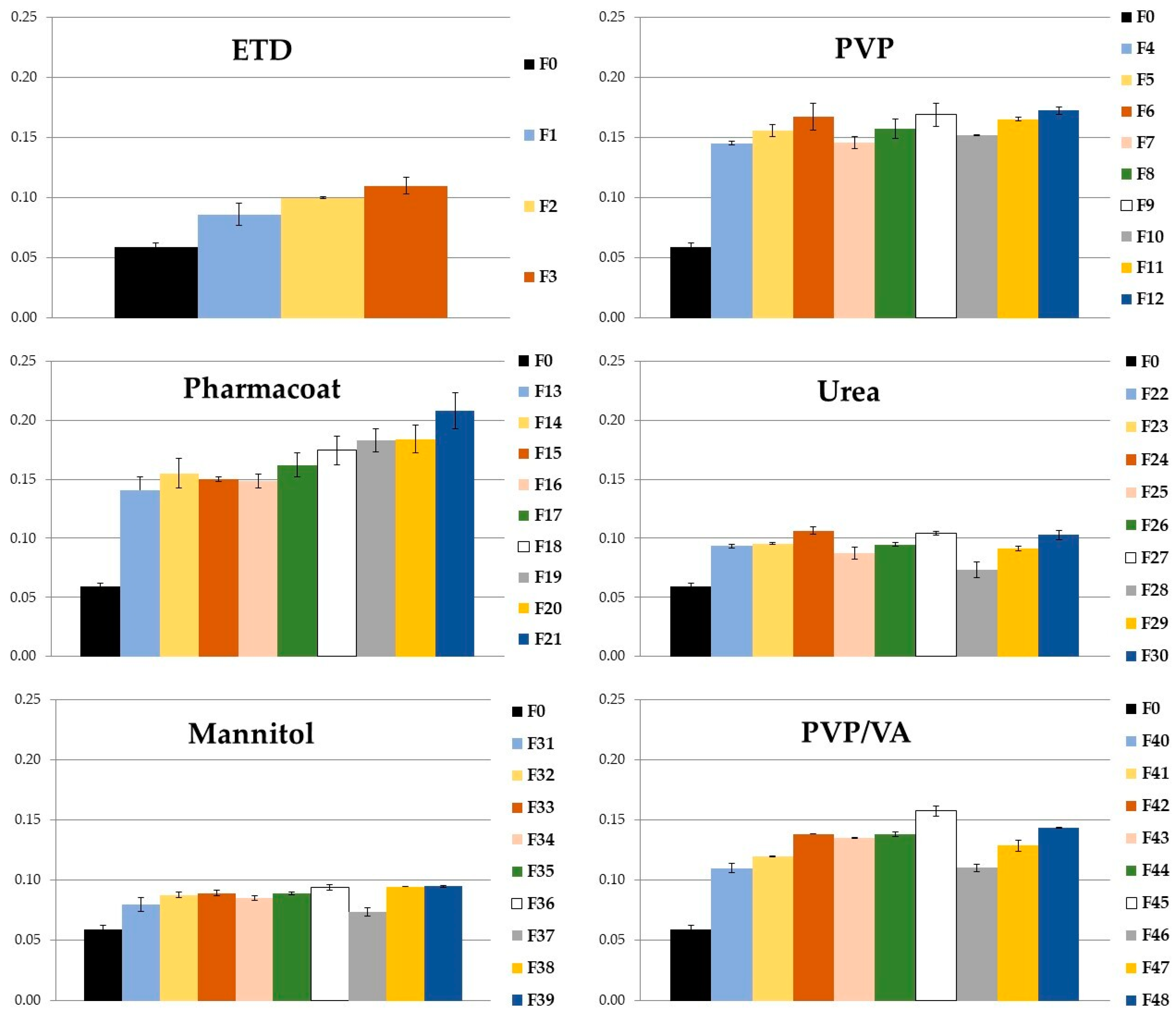
3.1.3. Statistical Analysis
3.2. 2nd and 3rd Generation of ETD-SDs
3.2.1. Drug Content and Solubility of ETD
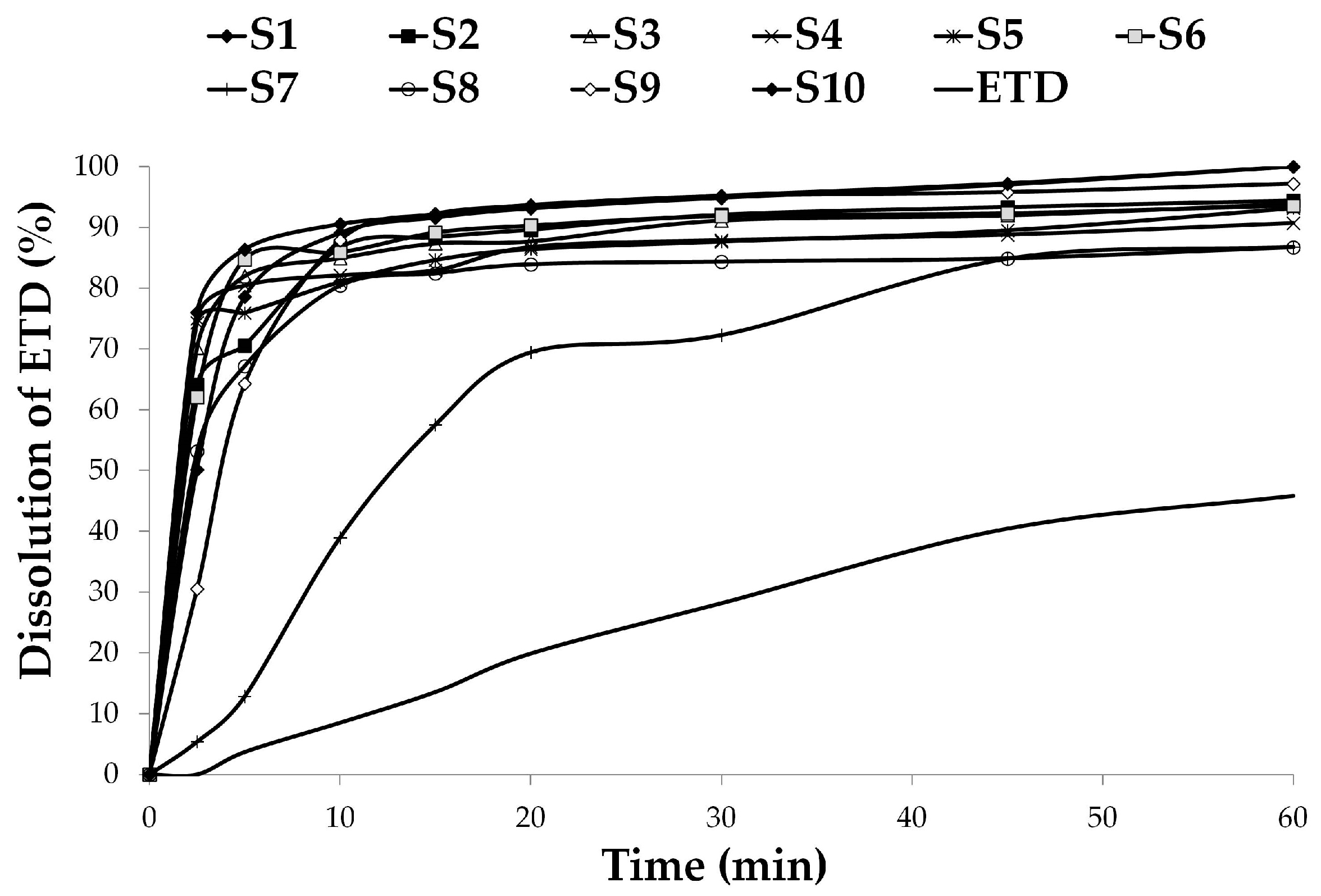
3.2.2. Dissolution Studies
3.2.3. Effect of Carriers and Poloxamer on SD Features—Statistical Analysis
3.2.4. Morphological Evaluation
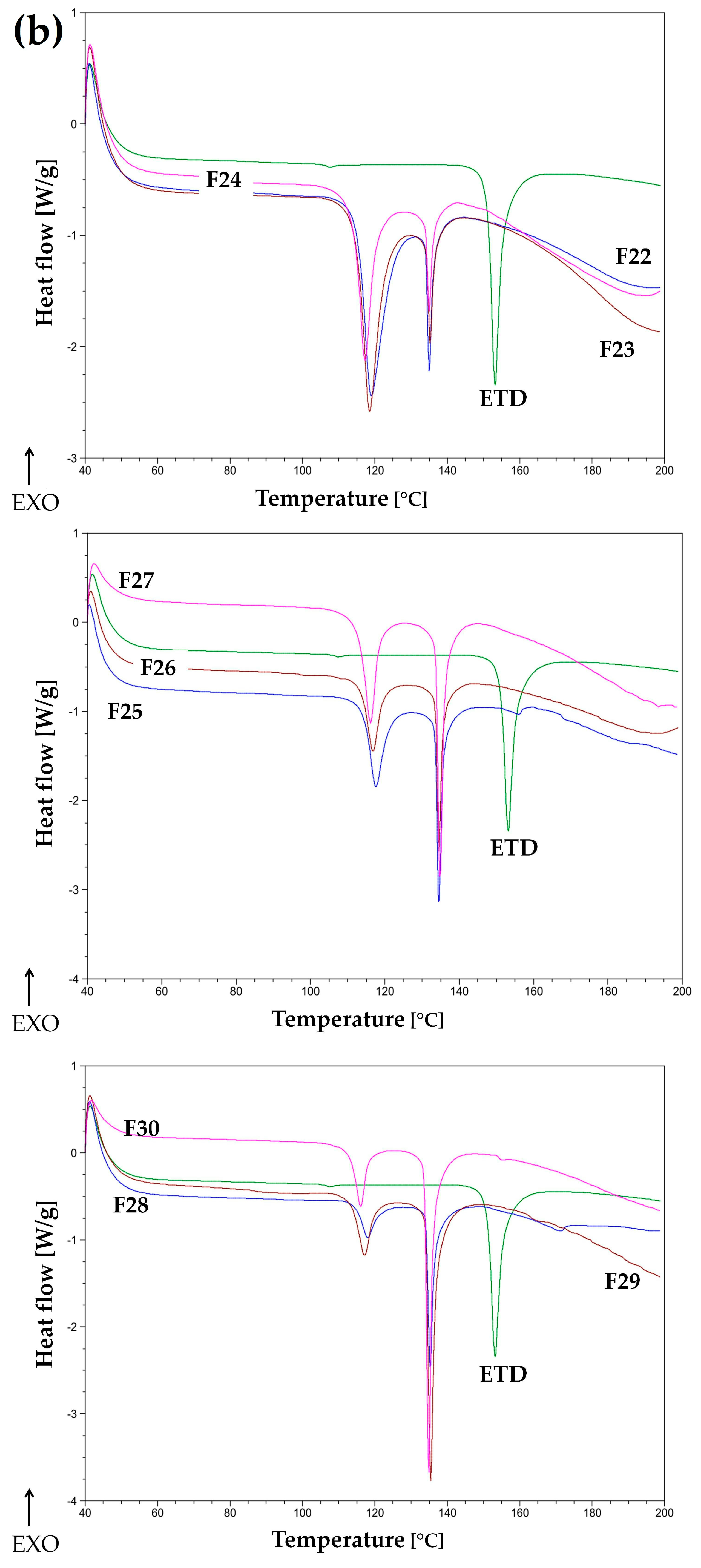
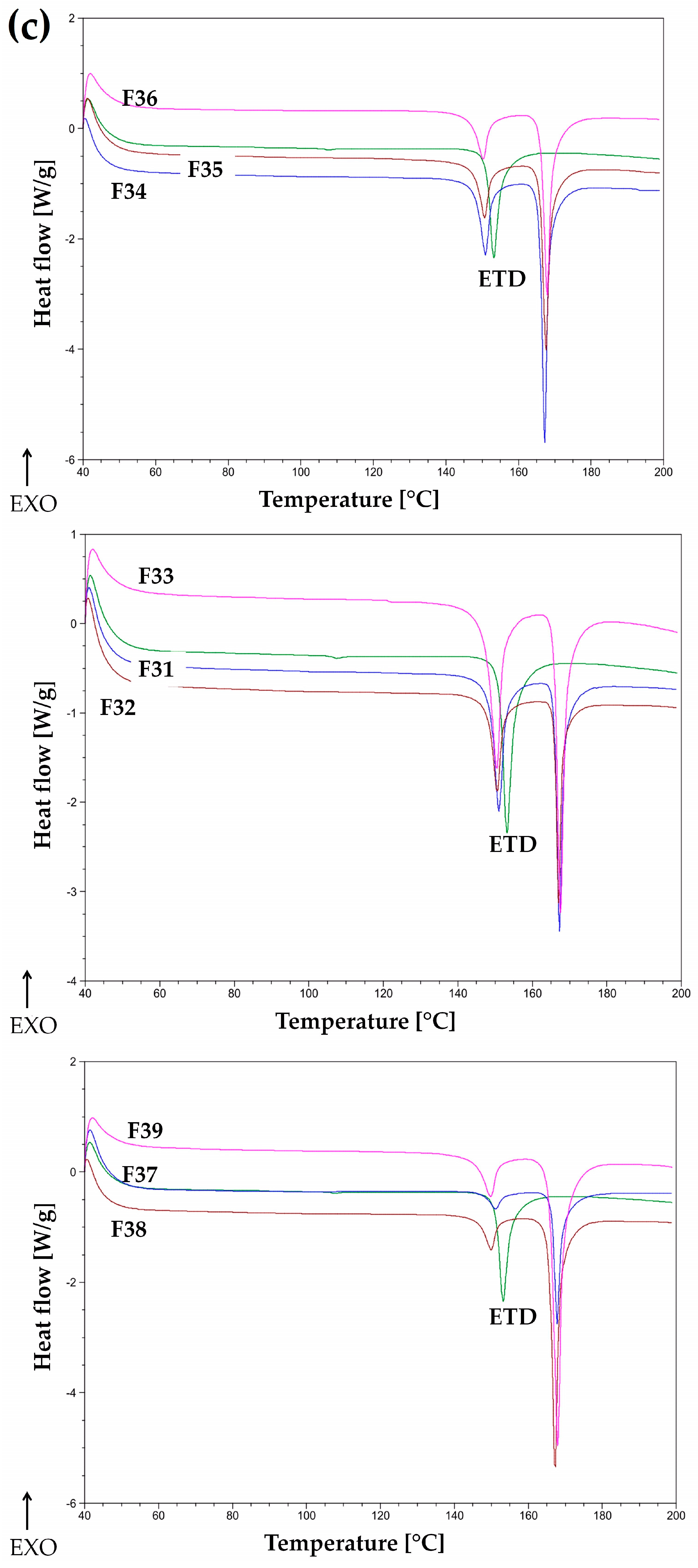
3.2.5. Thermal Analysis
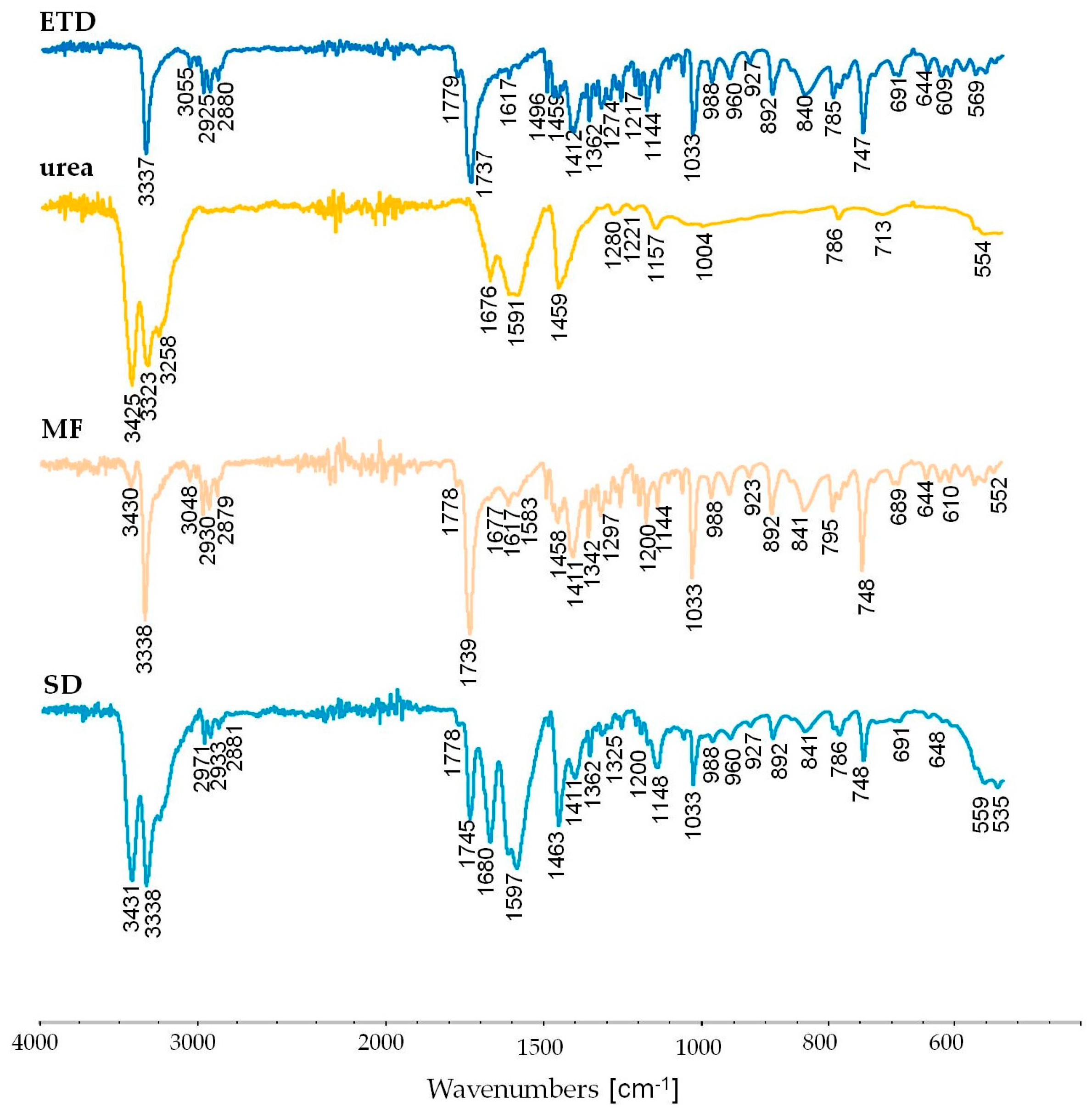
3.2.6. Attenuated Total Reflectance Fourier Transform Infrared Spectroscopy (ATR-FTIR) Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bhalani, D.V.; Nutan, B.; Kumar, A.; Singh Chandel, A.K. Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics. Biomedicines 2022, 10, 2055. [Google Scholar] [CrossRef] [PubMed]
- Reza Pahlevi, M.; Sopyan, I.; Gozali, D. Technique Development in Improving the Solubility of Poorly Water Soluble Drugs (BCS II and IV): A Review Study: Pengembangan Teknik Dalam Meningkatkan Kelarutan Obat Yang Larut Buruk Dalam Air (BCS Kelas II Dan IV): Studi Review. J. Farm. Galen. Galen. J. Pharm. E-J. 2023, 9, 147–164. [Google Scholar] [CrossRef]
- Da Silva, F.L.O.; Marques, M.B.D.F.; Kato, K.C.; Carneiro, G. Nanonization Techniques to Overcome Poor Water-Solubility with Drugs. Expert Opin. Drug Discov. 2020, 15, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Mehta, C.H.; Nayak, U.Y. Multiple Approaches for Achieving Drug Solubility: An in Silico Perspective. Drug Discov. Today 2020, 25, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.U.; Minhas, M.U.; Badshah, S.F.; Suhail, M.; Ahmad, A.; Ijaz, S. Overview of Nanoparticulate Strategies for Solubility Enhancement of Poorly Soluble Drugs. Life Sci. 2022, 291, 120301. [Google Scholar] [CrossRef]
- Vinarov, Z.; Katev, V.; Radeva, D.; Tcholakova, S.; Denkov, N.D. Micellar Solubilization of Poorly Water-Soluble Drugs: Effect of Surfactant and Solubilizate Molecular Structure. Drug Dev. Ind. Pharm. 2018, 44, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Malkawi, R.; Malkawi, W.I.; Al-Mahmoud, Y.; Tawalbeh, J. Current Trends on Solid Dispersions: Past, Present, and Future. Adv. Pharmacol. Pharm. Sci. 2022, 2022, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Abuzar, S.M.; Hyun, S.-M.; Kim, J.-H.; Park, H.J.; Kim, M.-S.; Park, J.-S.; Hwang, S.-J. Enhancing the Solubility and Bioavailability of Poorly Water-Soluble Drugs Using Supercritical Antisolvent (SAS) Process. Int. J. Pharm. 2018, 538, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.S.; Sahoo, J.; Agrawal, S.; Kumari, A. Solid Dispersions: A Technology for Improving Bioavailability. J. Anal. Pharm. Res. 2019, 8, 127–133. [Google Scholar] [CrossRef]
- Vo, C.L.-N.; Park, C.; Lee, B.-J. Current Trends and Future Perspectives of Solid Dispersions Containing Poorly Water-Soluble Drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef]
- Tran, P.; Pyo, Y.-C.; Kim, D.-H.; Lee, S.-E.; Kim, J.-K.; Park, J.-S. Overview of the Manufacturing Methods of Solid Dispersion Technology for Improving the Solubility of Poorly Water-Soluble Drugs and Application to Anticancer Drugs. Pharmaceutics 2019, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Tambosi, G.; Coelho, P.F.; Luciano, S.; Lenschow, I.C.S.; Zétola, M.; Stulzer, H.K.; Pezzini, B.R. Challenges to Improve the Biopharmaceutical Properties of Poorly Water-Soluble Drugs and the Application of the Solid Dispersion Technology. Matér. Rio Jan. 2018, 23. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble Drug Delivery Strategies: Review of Recent Advances and Business Prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef]
- Hallouard, F.; Mehenni, L.; Lahiani-Skiba, M.; Anouar, Y.; Skiba, M. Solid Dispersions for Oral Administration: An Overview of the Methods for Their Preparation. Curr. Pharm. Des. 2016, 22, 4942–4958. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.; Hasegawa, S.; Al-Obaidi, H.; Buckton, G. Investigation of Preparation Methods on Surface/Bulk Structural Relaxation and Glass Fragility of Amorphous Solid Dispersions. Int. J. Pharm. 2012, 422, 170–178. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid Dispersions as Strategy to Improve Oral Bioavailability of Poor Water Soluble Drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Van Den Mooter, G. Review: Physical Chemistry of Solid Dispersions. J. Pharm. Pharmacol. 2010, 61, 1571–1586. [Google Scholar] [CrossRef] [PubMed]
- Vippagunta, S.R.; Wang, Z.; Hornung, S.; Krill, S.L. Factors Affecting the Formation of Eutectic Solid Dispersions and Their Dissolution Behavior. J. Pharm. Sci. 2007, 96, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Shah, H.; Shah, V.; Bhutani, S.; Parikh, D.; Mehta, T. Dissolution Improvement of Nebivolol Hydrochloride Using Solid Dispersion Adsorbate Technique. Asian J. Pharm. 2015, 9, 49. [Google Scholar] [CrossRef]
- Babu, N.R.; Nagpal, D.; Ankola, D.; Awasthi, R. Evolution of Solid Dispersion Technology: Solubility Enhancement Using Hydroxypropyl Methylcellulose Acetate Succinate: Myth or Reality? ASSAY Drug Dev. Technol. 2022, 20, 149–163. [Google Scholar] [CrossRef]
- Bindhani, S.; Mohapatra, S. Recent Approaches of Solid Dispersion: A New Concept toward Oral Bioavailability Sabitri Bindhani*, Snehamayee Mohapatra. Asian J. Pharm. Clin. Res. 2018, 11, 72. [Google Scholar] [CrossRef]
- Tekade, A.R.; Yadav, J.N. A Review on Solid Dispersion and Carriers Used Therein for Solubility Enhancement of Poorly Water Soluble Drugs. Adv. Pharm. Bull. 2020, 10, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Vlachou, M.; Kikionis, S.; Siamidi, A.; Tragou, K.; Kapoti, S.; Ioannou, E.; Roussis, V.; Tsotinis, A. Fabrication and Characterization of Electrospun Nanofibers for the Modified Release of the Chronobiotic Hormone Melatonin. Curr. Drug Deliv. 2018, 16, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, R.; Löbmann, K.; Strachan, C.J.; Grohganz, H.; Rades, T. Emerging Trends in the Stabilization of Amorphous Drugs. Int. J. Pharm. 2013, 453, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.R.; Lakshman, Y.D.; Anand, V.S.K.; Sree, K.S.N.; Bhat, K.; Dengale, S.J. Overview of Extensively Employed Polymeric Carriers in Solid Dispersion Technology. AAPS PharmSciTech 2020, 21, 309. [Google Scholar] [CrossRef] [PubMed]
- BASF Pharma. Available online: https://pharma.basf.com/products/kollidon-25 (accessed on 15 March 2024).
- Van Duong, T.; Van Den Mooter, G. The Role of the Carrier in the Formulation of Pharmaceutical Solid Dispersions. Part I: Crystalline and Semi-Crystalline Carriers. Expert Opin. Drug Deliv. 2016, 13, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, K.; Tajber, L.; Sawicki, W. Physicochemical Properties of Direct Compression Tablets with Spray Dried and Ball Milled Solid Dispersions of Tadalafil in PVP-VA. Eur. J. Pharm. Biopharm. 2016, 109, 14–23. [Google Scholar] [CrossRef]
- Wlodarski, K.; Sawicki, W.; Haber, K.; Knapik, J.; Wojnarowska, Z.; Paluch, M.; Lepek, P.; Hawelek, L.; Tajber, L. Physicochemical Properties of Tadalafil Solid Dispersions—Impact of Polymer on the Apparent Solubility and Dissolution Rate of Tadalafil. Eur. J. Pharm. Biopharm. 2015, 94, 106–115. [Google Scholar] [CrossRef]
- DrugBank. Available online: https://go.drugbank.com/drugs/DB00749 (accessed on 10 March 2024).
- Liao, K.-F.; Cheng, K.-C.; Lin, C.-L.; Lai, S.-W. Etodolac and the Risk of Acute Pancreatitis. BioMedicine 2017, 7, 4. [Google Scholar] [CrossRef]
- Mrotek, E.; Dudziak, S.; Malinowska, I.; Pelczarski, D.; Ryżyńska, Z.; Zielińska-Jurek, A. Improved Degradation of Etodolac in the Presence of Core-Shell ZnFe2O4/SiO2/TiO2 Magnetic Photocatalyst. Sci. Total Environ. 2020, 724, 138167. [Google Scholar] [CrossRef]
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Poloxamer-407 (accessed on 20 March 2024).
- Czajkowska-Kośnik, A.; Szymańska, E.; Czarnomysy, R.; Jacyna, J.; Markuszewski, M.; Basa, A.; Winnicka, K. Nanostructured Lipid Carriers Engineered as Topical Delivery of Etodolac: Optimization and Cytotoxicity Studies. Materials 2021, 14, 596. [Google Scholar] [CrossRef] [PubMed]
- Madhavi, N.; Sudhakar, B.; Suresh Reddy, K.V.N.; Vijaya Ratna, J. Pharmacokinetic and Pharmacodynamic Studies of Etodolac Loaded Vesicular Gels on Rats by Transdermal Delivery. DARU J. Pharm. Sci. 2018, 26, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Bachhav, D.G.; Khadabadi, S.S.; Deore, L.P. Development and Validation of HPLC Method for Estimation of Etodolac in Rat Plasma. Austin J. Anal. Pharm. Chem. 2016, 3, 1061. [Google Scholar]
- Cherukuri, S.; Batchu, U.; Mandava, K.; Cherukuri, V.; Ganapuram, K. Formulation and Evaluation of Transdermal Drug Delivery of Topiramate. Int. J. Pharm. Investig. 2017, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Singh, S.K.; Singh, S.; Sheth, N.R.; Gendle, R. Development and Characterization of Curcumin Loaded Transfersome for Transdermal Delivery. J. Pharm. Sci. Res. 2009, 1, 71–80. [Google Scholar]
- Özer, Ö.; Mutlu, B.; Tömek, S. Development and in Vitro Evaluation of Topically Applied Cinnamic Acid Formulations. Pharmazie 2007, 62, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An Add-In Program for Modeling and Comparison of Drug Dissolution Profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Fouad, S.A.; Malaak, F.A.; El-Nabarawi, M.A.; Abu Zeid, K.; Ghoneim, A.M. Preparation of Solid Dispersion Systems for Enhanced Dissolution of Poorly Water Soluble Diacerein: In-Vitro Evaluation, Optimization and Physiologically Based Pharmacokinetic Modeling. PLoS ONE 2021, 16, e0245482. [Google Scholar] [CrossRef]
- Karolewicz, B.; Górniak, A.; Owczarek, A.; Nartowski, K.; Żurawska-Płaksej, E.; Pluta, J. Solid Dispersion in Pharmaceutical Technology. Part II. The Methods of Analysis of Solid Dispersions and Examples of Their Application. Polim. Med. 2012, 42, 97–107. [Google Scholar]
- Huang, B.B.; Liu, D.X.; Liu, D.K.; Wu, G. Application of Solid Dispersion Technique to Improve Solubility and Sustain Release of Emamectin Benzoate. Molecules 2019, 24, 4315. [Google Scholar] [CrossRef]
- Suknuntha, K.; Khumpirapang, N.; Tantishaiyakul, V.; Okonogi, S. Solubility and Physical Stability Enhancement of Loratadine by Preparation of Co-Amorphous Solid Dispersion with Chlorpheniramine and Polyvinylpyrrolidone. Pharmaceutics 2023, 15, 2558. [Google Scholar] [CrossRef]
- Budiman, A.; Handini, A.L.; Muslimah, M.N.; Nurani, N.V.; Laelasari, E.; Kurniawansyah, I.S.; Aulifa, D.L. Amorphous Solid Dispersion as Drug Delivery Vehicles in Cancer. Polymers 2023, 15, 3380. [Google Scholar] [CrossRef] [PubMed]
- Talik, P.; Hubicka, U. Differential Scanning Calorimetry (DSC) in the Study of Potential Incompatibilities between Fluconazole and Selected Pharmaceutical Excipients. Med. Int. Rev. 2018, 28, 82–90. [Google Scholar]
- Iijima, M.; Hatakeyama, T.; Hatakeyama, H. DSC and TMA Studies of Polysaccharide Physical Hydrogels. Anal. Sci. 2021, 37, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Janovszky, D.; Sveda, M.; Sycheva, A.; Kristaly, F.; Zámborszky, F.; Koziel, T.; Bala, P.; Czel, G.; Kaptay, G. Amorphous Alloys and Differential Scanning Calorimetry (DSC). J. Therm. Anal. Calorim. 2022, 147, 7141–7157. [Google Scholar] [CrossRef]
- Bhujbal, S.V.; Mitra, B.; Jain, U.; Gong, Y.; Agrawal, A.; Karki, S.; Taylor, L.S.; Kumar, S.; Zhou, Q. Pharmaceutical Amorphous Solid Dispersion: A Review of Manufacturing Strategies. Acta Pharm. Sin. B 2021, 11, 2505–2536. [Google Scholar] [CrossRef] [PubMed]
- Onija, O.; Borodi, G.; Kacso, I.; Pop, M.N.; Dadarlat, D.; Bratu, I.; Jumate, N.; Lazar, M.D. Preparation and Characterization of Urea-Oxalic Acid Solid Form. AIP Conf. Proc. 2012, 1425, 35–38. [Google Scholar] [CrossRef]
- Zhu, N.; Qian, F.; Xu, X.; Wang, M.; Teng, Q. Thermogravimetric Experiment of Urea at Constant Temperatures. Materials 2021, 14, 6190. [Google Scholar] [CrossRef]
- Khodaverdi, E.; Khalili, N.; Zangiabadi, F.; Homayouni, A. Preparation, Characterization and Stability Studies of Glassy Solid Dispersions of Indomethacin Using PVP and Isomalt as Carriers. Iran J. Basic Med Sci. 2012, 15, 820–832. [Google Scholar]
- Leyk, E.; Wesolowski, M. Interactions between Paracetamol and Hypromellose in the Solid State. Front. Pharmacol. 2019, 10, 14. [Google Scholar] [CrossRef]
- Pandi, P.; Bulusu, R.; Kommineni, N.; Khan, W.; Singh, M. Amorphous Solid Dispersions: An Update for Preparation, Characterization, Mechanism on Bioavailability, Stability, Regulatory Considerations and Marketed Products. Int. J. Pharm. 2020, 586, 119560. [Google Scholar] [CrossRef] [PubMed]
- Zajc, N.; Obreza, A.; Bele, M.; Srčič, S. Physical Properties and Dissolution Behaviour of Nifedipine/Mannitol Solid Dispersions Prepared by Hot Melt Method. Int. J. Pharm. 2005, 291, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Li, J.; Wang, H.; Pan, H.; Li, T.; Bu, T.; Zhou, W.; Zhang, X. Role of Polymers in the Physical and Chemical Stability of Amorphous Solid Dispersion: A Case Study of Carbamazepine. Eur. J. Pharm. Sci. 2022, 169, 106086. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, K.; Sawicki, W.; Kozyra, A.; Tajber, L. Physical Stability of Solid Dispersions with Respect to Thermodynamic Solubility of Tadalafil in PVP-VA. Eur. J. Pharm. Biopharm. 2015, 96, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Wade, A.; Weller, P.J. Handbook of Pharmaceutical Excipients, 5th ed.; Pharmaceutical Press and American Pharmacists Association: Washington, DC, USA, 2006. [Google Scholar]
- Vasconcelos, T.; Prezotti, F.; Araújo, F.; Lopes, C.; Loureiro, A.; Marques, S.; Sarmento, B. Third-Generation Solid Dispersion Combining Soluplus and Poloxamer 407 Enhances the Oral Bioavailability of Resveratrol. Int. J. Pharm. 2021, 595, 120245. [Google Scholar] [CrossRef] [PubMed]
- Rosiak, N.; Wdowiak, K.; Tykarska, E.; Cielecka-Piontek, J. Amorphous Solid Dispersion of Hesperidin with Polymer Excipients for Enhanced Apparent Solubility as a More Effective Approach to the Treatment of Civilization Diseases. Int. J. Mol. Sci. 2022, 23, 15198. [Google Scholar] [CrossRef]
- Huang, Y.; Zhao, X.; Zu, Y.; Wang, L.; Deng, Y.; Wu, M.; Wang, H. Enhanced Solubility and Bioavailability of Apigenin via Preparation of Solid Dispersions of Mesoporous Silica Nanoparticles. Iran. J. Pharm. Res. 2019, 18, 168–182. [Google Scholar] [PubMed]
- Wdowiak, K.; Tajber, L.; Miklaszewski, A.; Cielecka-Piontek, J. Sweeteners Show a Plasticizing Effect on PVP K30—A Solution for the Hot-Melt Extrusion of Fixed-Dose Amorphous Curcumin-Hesperetin Solid Dispersions. Pharmaceutics 2024, 16, 659. [Google Scholar] [CrossRef]
- Bhujbal, S.V.; Pathak, V.; Zemlyanov, D.Y.; Taylor, L.S.; Zhou, Q. Physical Stability and Dissolution of Lumefantrine Amorphous Solid Dispersions Produced by Spray Anti-Solvent Precipitation. J. Pharm. Sci. 2021, 110, 2423–2431. [Google Scholar] [CrossRef]
- Woo, J.-H.; Ngo, H.V.; Nguyen, H.D.; Gil, M.-C.; Park, C.; Park, J.-B.; Cui, J.-H.; Cao, Q.-R.; Lee, B.-J. Polyelectrolyte-Based Solid Dispersions for Enhanced Dissolution and pH-Independent Controlled Release of Sildenafil Citrate. Heliyon 2023, 9, e23091. [Google Scholar] [CrossRef] [PubMed]
- Aboutaleb, A.; Abdel-Rahman, S.; Ahmed, M.; Younis, M. Improvement of Domperidone Solubility and Dissolution Rate by Dispersion in Various Hydrophilic Carriers. J. Appl. Pharm. Sci. 2016, 6, 133–139. [Google Scholar] [CrossRef]
- Costa, P.; Sousa Lobo, J.M. Modeling and Comparison of Dissolution Profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Alam, S.; Paul, P. Development and Evaluation of Sustained Release Matrix Tablets of Indapamide Using Methocel K15M CR. J. Appl. Pharm. Sci. 2013, 3, 85–90. [Google Scholar]
- Szafraniec, J.; Antosik, A.; Knapik-Kowalczuk, J.; Chmiel, K.; Kurek, M.; Gawlak, K.; Odrobińska, J.; Paluch, M.; Jachowicz, R. The Self-Assembly Phenomenon of Poloxamers and Its Effect on the Dissolution of a Poorly Soluble Drug from Solid Dispersions Obtained by Solvent Methods. Pharmaceutics 2019, 11, 130. [Google Scholar] [CrossRef] [PubMed]
- Shah, T.J.; Amin, A.F.; Parikh, J.R.; Parikh, R.H. Process Optimization and Characterization of Poloxamer Solid Dispersions of a Poorly Water-Soluble Drug. AAPS PharmSciTech 2007, 8, E18–E24. [Google Scholar] [CrossRef]
- Eloy, J.O.; Marchetti, J.M. Solid Dispersions Containing Ursolic Acid in Poloxamer 407 and PEG 6000: A Comparative Study of Fusion and Solvent Methods. Powder Technol. 2014, 253, 98–106. [Google Scholar] [CrossRef]
- Wojcik-Pastuszka, D.; Krzak, J.; Macikowski, B.; Berkowski, R.; Osiński, B.; Musiał, W. Evaluation of the Release Kinetics of a Pharmacologically Active Substance from Model Intra-Articular Implants Replacing the Cruciate Ligaments of the Knee. Materials 2019, 12, 1202. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Murthy, P.N.; Nath, L. Kinetic Modeling on Drug Release from Controlled Drug Delivery Systems. Acta Pol Pharm. 2010, 67, 217–223. [Google Scholar]
- Kumar, A.; Kumar, J. Solid Dispersion Techniques: A Review. Int. J. Res. Eng. Sci. Manag. 2021, 4, 104–111. [Google Scholar]
- Sanklecha, V.M. A Systematic Review on Solid Dispersion: Enhancing the Solubility of Poorly Soluble Drug. Austin J. Nanomed. Nanotechno. 2020, 8, 1059. [Google Scholar]
- Alshehri, S.; Imam, S.S.; Altamimi, M.A.; Hussain, A.; Shakeel, F.; Elzayat, E.; Mohsin, K.; Ibrahim, M.; Alanazi, F. Enhanced Dissolution of Luteolin by Solid Dispersion Prepared by Different Methods: Physicochemical Characterization and Antioxidant Activity. ACS Omega 2020, 5, 6461–6471. [Google Scholar] [CrossRef] [PubMed]
- Annepogu, H.; Ahad, H.A.; Nayakanti, D. Determining the Best Poloxamer Carrier for Thiocolchicoside Solid Dispersions. Turk. J. Pharm. Sci. 2020, 17, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Ei-Badry, M.; Hassan, M.A.; Ibrahim, M.A.; Elsaghir, H. Performance of Poloxamer 407 as Hydrophilic Carrier In The Binary Mixtures With Niemsulide. Farmacia 2013, 61, 1137–1150. [Google Scholar]
- Ford, J.L.; Mann, T.E. Fast-Scan DSC and Its Role in Pharmaceutical Physical Form Characterisation and Selection. Adv. Drug Deliv. Rev. 2012, 64, 422–430. [Google Scholar] [CrossRef]
- Rosiak, N.; Tykarska, E.; Cielecka-Piontek, J. The Study of Amorphous Kaempferol Dispersions Involving FT-IR Spectroscopy. Int. J. Mol. Sci. 2023, 24, 17155. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ETD-SD | Ratio of ETD:Carrier | Time of Milling (min) |
|---|---|---|
| F1, F2, F3 | 1:0 | 15, 30, 60 |
| PVP | ||
| F4, F5, F6 | 2:1 | 15, 30, 60 |
| F7, F8, F9 | 1:1 | 15, 30, 60 |
| F10, F11, F12 | 1:2 | 15, 30, 60 |
| Pharmacoat | ||
| F13, F14, F15 | 2:1 | 15, 30, 60 |
| F16, F17, F18 | 1:1 | 15, 30, 60 |
| F19, F20, F21 | 1:2 | 15, 30, 60 |
| Urea | ||
| F22, F23, F24 | 2:1 | 15, 30, 60 |
| F25, F26, F27 | 1:1 | 15, 30, 60 |
| F28, F29, F30 | 1:2 | 15, 30, 60 |
| Mannitol | ||
| F31, F32, F33 | 2:1 | 15, 30, 60 |
| F34, F35, F36 | 1:1 | 15, 30, 60 |
| F37, F38, F39 | 1:2 | 15, 30, 60 |
| PVP/VA | ||
| F40, F41, F42 | 2:1 | 15, 30, 60 |
| F43, F44, F45 | 1:1 | 15, 30, 60 |
| F46, F47, F48 | 1:2 | 15, 30, 60 |
| ETD-SD | ETD (g) | Polymer (g) | Poloxamer (g) |
|---|---|---|---|
| Pharmacoat | |||
| S1 | 1 | 1 | 1 |
| S2 | 1 | 0.5 | 1.5 |
| PVP | |||
| S3 | 1 | 1 | 1 |
| S4 | 1 | 0.5 | 1.5 |
| PVP/VA | |||
| S5 | 1 | 1 | 1 |
| S6 | 1 | 0.5 | 1.5 |
| Pharmacoat | |||
| S7 | 1 | 2 | 0 |
| PVP | |||
| S8 | 1 | 2 | 0 |
| PVP/VA | |||
| S9 | 1 | 2 | 0 |
| Poloxamer 407 | |||
| S10 | 1 | 0 | 2 |
| Formulation | Solubility in Water (mg/mL) * | Solubility in Phosphate Buffer pH 7.4 (mg/mL) ** |
|---|---|---|
| S1 (Pharmacoat:Poloxamer = 1:1) | 0.19 a ± 0.01 | 1.69 ± 0.02 |
| S2 (Pharmacoat:Poloxamer = 0.5:1.5) | 0.25 b ± 0.00 | 1.77 ± 0.03 |
| S3 (PVP:Poloxamer = 1:1) | 0.19 ca ± 0.01 | 1.66 ± 0.06 |
| S4 (PVP:Poloxamer = 0.5:1.5) | 0.16 dc ± 0.01 | 1.64 ± 0.13 |
| S5 (PVP/VA:Poloxamer = 1:1) | 0.21 eac ± 0.01 | 1.77 ± 0.07 |
| S6 (PVP/VA:Poloxamer = 0.5:1.5) | 0.22 fbe ± 0.01 | 1.86 ± 0.04 |
| S7 (Pharmacoat) | 0.16 gd ± 0.01 | 1.73 ± 0.07 |
| S8 (PVP) | 0.12 h ± 0.01 | 1.68 ± 0.03 |
| S9 (PVP/VA) | 0.14 idgh ± 0.01 | 1.73 ± 0.02 |
| S10 (Poloxamer) | 0.26 jb ± 0.01 | 1.82 ± 0.11 |
| Formulation | Dissolution Parameters | Similarity Factor, f2 | |
|---|---|---|---|
| DE (%) | MDT (min) | ||
| S1 | 0.90 | 5.87 | 9.7 |
| S2 | 0.87 | 4.58 | 10.4 |
| S3 | 0.87 | 4.35 | 9.9 |
| S4 | 0.85 | 4.04 | 10.5 |
| S5 | 0.85 | 5.42 | 10.6 |
| S6 | 0.88 | 3.80 | 9.9 |
| S7 | 0.65 | 15.15 | 22.8 |
| S8 | 0.80 | 4.57 | 12.8 |
| S9 | 0.88 | 5.72 | 10.9 |
| S10 | 0.92 | 4.72 | 8.4 |
| ETD | 0.26 | 25.51 | - |
| Formulation | Correlation Coefficient (R2) | K * (min−1) | ||
|---|---|---|---|---|
| Zero Order | First Order | Higuchi Model | ||
| S1 | 0.632 | 0.995 | 0.821 | 0.276 |
| S2 | 0.599 | 0.984 | 0.795 | 0.296 |
| S3 | 0.545 | 0.986 | 0.744 | 0.404 |
| S4 | 0.514 | 0.976 | 0.713 | 0.437 |
| S5 | 0.553 | 0.970 | 0.745 | 0.393 |
| S6 | 0.549 | 0.991 | 0.752 | 0.360 |
| S7 | 0.885 | 0.985 | 0.958 | 0.048 |
| S8 | 0.596 | 0.975 | 0.796 | 0.209 |
| S9 | 0.668 | 0.995 | 0.847 | 0.184 |
| S10 | 0.542 | 0.993 | 0.739 | 0.509 |
| ETD | 0.989 | 0.997 | 0.969 | 0.011 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czajkowska-Kośnik, A.; Misztalewska-Turkowicz, I.; Wilczewska, A.Z.; Basa, A.; Winnicka, K. Solid Dispersions Obtained by Ball Milling as Delivery Platform of Etodolac, a Model Poorly Soluble Drug. Materials 2024, 17, 3923. https://doi.org/10.3390/ma17163923
Czajkowska-Kośnik A, Misztalewska-Turkowicz I, Wilczewska AZ, Basa A, Winnicka K. Solid Dispersions Obtained by Ball Milling as Delivery Platform of Etodolac, a Model Poorly Soluble Drug. Materials. 2024; 17(16):3923. https://doi.org/10.3390/ma17163923
Chicago/Turabian StyleCzajkowska-Kośnik, Anna, Iwona Misztalewska-Turkowicz, Agnieszka Zofia Wilczewska, Anna Basa, and Katarzyna Winnicka. 2024. "Solid Dispersions Obtained by Ball Milling as Delivery Platform of Etodolac, a Model Poorly Soluble Drug" Materials 17, no. 16: 3923. https://doi.org/10.3390/ma17163923